
































关于高效液相首选等度洗脱,流动相比例越简单越好,能不用缓冲盐就不用,洗脱方法越简单越好、色谱柱越常见越好,但是有的时候用单一比例的流动相或者是常见色谱柱是无法满足很多杂质的分离,这时就需重新开发色谱分离方式。下面为一些分析条件开发的个人的经验,希望给新入行的朋友一些实验灵感。
1、确定目标化合物性质
首先需要明白待分离杂质的一些性质,比如说分子量、结构式(主要看有哪种基团),以及该种物质在哪种有机溶剂中易容。结构式对于分子极性大小的预测以及判定该物质是否容易水解有非常重要的作用。例如我们常见的-、-NHR、-、-COOH、-0H等都是比较常见的极性基团;而常见的苯环、-CH=、--等都是常见的非极性基团。根据经验可以大致的判断一下物质的极性,预估一下在常用的色谱柱上是否保留,再结合杂质在各种常见洗脱溶剂中的溶解性,例如在甲醇、乙腈、正己烷中哪个比较易溶,选择适合用于洗脱的有机流动相,从而确定方法开发时是用正相色谱柱或者是反向色谱柱做出粗略判断。
2、确定正反相洗脱方式
确定洗脱方式后,反向的话个人推荐先用60%的乙腈或者是甲醇,先去看一下你需要分离的几种物质能否被分开,(正相洗脱方式思路相近)。在能分开的前提下,再看一下最后一个峰的出峰时间,如果时间比较长的话,再根据前几个峰的分离度确定是否需要加大或者缩小有机相的比例。我们常见的洗脱分离方式为反相分离,而反相洗脱方式又分为两种体系,一种是甲醇-水体系,另外一种是乙腈-水体系。
具体是选择哪种反相洗脱体系需要根据以下条件来决定:
A、乙腈的洗脱能力要优于甲醇,在同等浓度、样品下乙腈-水体系的目标峰出峰要快于甲醇-水体系。
B、甲醇溶于水是会放出热量的,乙腈溶于水是会吸热的,在同等方式的梯度洗脱方式下,在两种流动相互相混合时由于分子热运动的原因,甲醇溶于水比乙腈溶于水时的分子热运动剧烈,这样两种流动相的混合会更加均匀,反映在基线上的表现为基线更加的平滑,且梯度峰要更少。
C、甲醇在市面上的售价要低于乙腈(如果财大气粗的公司可以忽略这一条)。
D、甲醇的分子量大于乙腈,在同一色谱柱、相同浓度的有机相浓度下,乙腈-水体系的柱压更小。
E、甲醇的截止吸收波长在210nm附近,而乙腈的截止吸收波长在190nm附近,在检测波长远离210nm的时候选择甲醇或是乙腈-水体系主要看其它影响因素。但是测定波长在210nm附近时,由于甲醇的截止吸收波长大于乙腈,除了流动相不同外,相同的测定条件下,乙腈-水体系测定的样品响应值更高,从而能够得到更小样品检出限,此时建议优选乙腈-水体系。
图2-1以甲醇-水为体系的系统适用性图谱
图2-2 以乙腈-水为体系的系统适用性图谱
上述两个图中图2-2比图2-1多加入一种杂质,即图2中的峰5,其余杂质的种类不变、出峰位置不变,上述两图主要看图谱的基线与各杂质的出峰位置。
3、确定色谱柱、流速、柱温
实验室分析中最常用的就是类型的色谱柱,一般为5μm0.46×250mm或者是0.46×150mm,在试验前期如果在没有参考资料的情况下,首选最为普通的 5μm0.46×250mm的色谱柱试一下;如果发现用5μm0.46×250mm的色谱柱去分离多种杂质时,有分叉峰出现或是理论塔板数比较低时则提示我们这种类型的色谱柱对该物质的分离是不太好的,需要我们更换柱效更高的色谱柱,通常选择粒径、填料更小的色谱柱,这个需要根据实际情况,进行试验后确定适用于检测的色谱柱。
而流速的选择通常是要根据色谱柱的具体类型,最高的耐受压力来决定,常用的色谱柱一般流速为1.0mL/min,其实流量对杂质分离的影响比较小。
柱温首选室温,柱温的升高会影响杂质的出峰快慢,理论上柱温越高,各种物质的出峰会越快,同时色谱柱的柱压也会越小,但是由于温度比较高会导致色谱柱中填料的健合相容易被流动相冲刷下来,导致色谱柱不可逆的柱效下降。因此色谱柱温度不宜过高,一般温度设定为25~35℃,当然也有特殊的品种有的需要柱温在40℃甚至更高。
4、梯度洗脱程序的筛选
在选择好用正反相洗脱方式、确定用的色谱柱、柱温、流速后,无论如何调整梯度及流动相比例都无法将两种杂质分离开后,那么就需要用到梯度洗脱方式。梯度洗脱是一种在不同的时间加入不同有机比例流动相的一种洗脱方式。与传统的等度相洗脱方式比具有以下优点:
A、能够增加两种物质的分离度,通过不同时间段加入有机相浓度的不同,使杂质在不同时间段受到有机相洗脱的浓度不同,一般原则是逐步增加有机相的比例,从而使难被洗脱出来的杂质加快出峰。
B、由于可以调整有机相加入的浓度,与等度洗脱相比,可以缩短分析时间,还可以一次性将等度洗脱分不开的几种物质分开,从而用一种分析方法分析多种物质,降低成本。
梯度洗脱程序的摸索过程,需要遵循以下原则:
原则一:不使用单一流动相
初级实验者梯度洗脱的时候,流动相A一般为缓冲盐,流动相B一般为纯的有机相。但是高级实验者使用的A相往往不单纯使用缓冲盐,而是要加入一定比例的有机相,原因有两个。第一是加入最少5%的甲醇或者乙腈可以防止流动相长菌,减少频繁的过滤流动相这一操作。第二是加入一定比例的有机相可以减小在仪器进行流动相A、B相互混合时造成的基线波动,使基线更加平滑。至于流动相B到底是否是选择纯的有机相还是选择一定比例的有机相例如(80%、90%的甲醇或乙腈)要根据实验去做选择。本人推荐用一定比例的有机相为流动相B。因为在加入纯的乙腈或者甲醇等有机相时,由于两相在混合时会有吸热或放热的反应,会有很大的基线波动,用一定比例的有机相作为B相可以有效地减小流动相冲击效应。
原则二:有机相加入梯度梯度尽量缓
以下为笔者自己的一个实验项目,在进行梯度方法摸索时的一个案例,直接上图。
图4-1 梯度25图谱
梯度方法25洗脱程序
图4-2 梯度26图谱
梯度方法26洗脱程序
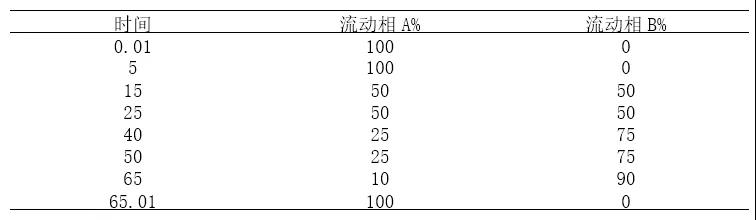
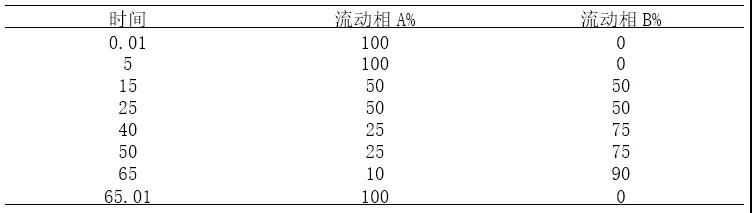
梯度方法27洗脱方式
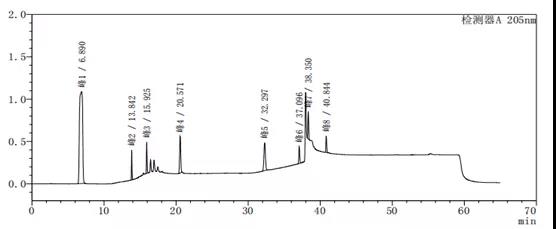
图4-3 梯度27图谱
梯度方法27洗脱方式
通过上述图谱、梯度洗脱程序的比较可以看出,缓慢的加入有机相可以使梯度峰更加平滑,使各杂质峰的分离度更好。
关于梯度鬼峰的出现,如果纯的缓冲盐相与有机溶剂相相互混合时,会有很大的互溶冲击效应的出现,这时候即使安装市面上的比较常用的鬼峰捕集小柱,基线依然不是很平滑;所以我推荐用混合一定比例的有机相与盐相的溶液作为混合盐相,将混有一定比例水的有机相的混合液作为梯度洗脱的有机相,配合鬼峰捕集小柱的使用可以使基线更加平滑。
5、缓冲盐的选择
首先要明白为什么要加入缓冲盐,缓冲盐的作用之一是为了增加流动相的缓冲能力。一般常见的无机缓冲盐(磷酸二氢钾、磷酸氢二钾、磷酸二氢氨、乙酸铵等)的作用是为了调整流动相的缓冲能力;何为缓冲能力?缓冲能力是指在其他溶液或者物质加入到流动相后,流动相pH变化程度的大小,流动相的pH变化越小证明流动相的缓冲能力越强,反之越弱。在一些标准上可以看到同一流动相的配置过程中需要使用的缓冲盐有两种,这是因为使用复合盐作为流动相比使用单一的盐作为流动相的缓冲能力强。有些待测主成分在中性或者碱性的环境中容易水解,例如带-OH的基团,容易在中性、碱性环境中水解,在图谱上表现为双头峰。为防止这种类型的水解,可以加入磷酸盐用磷酸调节pH至酸性环境,从而抑制水解。
加入缓冲盐的另外作用是为了增加待测成分的保留时间,通常带有氨基的物质或者氨根的物质如-、-NHR、-等基团时,为了增加该物质的保留,可以添加-(硫酸基)的离子对试剂如戊烷磺酸钠、辛烷磺酸钠,以增强保留。
一般来说缓冲盐是不会出峰的,我曾经用pH调为3.0的水为流动相与用磷酸二氢钾为缓冲盐pH调为3.0的水溶液作为流动相,两者图谱出来的梯度峰吻合度非常好,所以各位同仁不必担心缓冲盐会造成梯度峰。造成梯度峰的直接原因是流动相混合冲击。
6、检测波长的选择
关于检测波长的选择,需要结合多个杂质的紫外吸收图谱去综合考量,最好选择紫外吸收特征图谱上波峰处的波长作为检测波长,但是也要兼顾其它杂质在该波长下是否有紫外吸收。
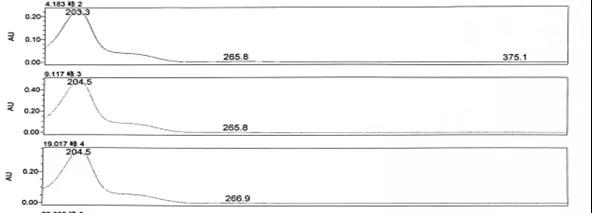
图6-1 杂质的紫外吸收特征图谱
以上述图谱中三个杂质为例,可以看出3个杂质的最大吸收分别为203.3、204.5、204.5nm,同时在205~230nm这个范围内也可以作为次选波长。所以综合考虑该三个杂质的检测波长可以暂定为205nm。
波长选择需要注意的点:190~210nm这个范围内的波长慎重选择,该波段的波长属于低波段,受到的干扰比较多,尤其是受流动相中试剂的纯度影响较大。如果遇到试剂不纯或者水质不好时,就会有一个类似梯度峰的“峰”出现,此类峰往往比较平滑,并不像真正杂质的峰那样尖锐,同时表现为无论如何调整梯度洗脱程序,始终在特定的位置“出峰”。如果该“峰”恰巧影响杂质出峰,那么消除起来就非常难了。笔者曾经遇到的一个项目就被这种峰纠缠了好久,后来通过更换更高级别的试剂、更纯净的水、多次冲洗仪器、色谱柱,逐步排除后才解决问题,我曾试验过,用DAD检测器选用265nm、225nm、205nm分别作为检测波长,该“峰”在265nm、225nm不会出现,所以205这个波长慎重选择。
7、溶剂的选择
如果用等度洗脱,首选用流动相作为溶剂,一般直接用流动相为稀释剂已经足够满足日常分析要求。但对于梯度洗脱方式,最好选择与初始梯度浓度相近溶液作为溶剂;但是不管是等度还是梯度洗脱,都要需要考虑以下几点;第一是溶剂对样品的溶解能力,如果所选的溶剂对样品的溶解能力不足,轻则不出峰,重则堵仪器、色谱柱。第二是需要考虑溶剂出峰是否会对目标峰的分离造成影响,如果溶剂峰影响目标峰出峰,那么所选的溶剂肯定是不合适的。对于梯度洗脱溶剂的选择,可以通过逐步调节溶剂中有机相与盐相的比例,观察图谱来选择合适的溶剂,同时要注意溶剂的pH、缓冲能力要尽可能的与流动相相当。
我们一直说的溶剂峰到底是个什么鬼?很多同仁做了很多年实验也没有真正的搞清楚,溶剂峰是由于溶剂中有机相与初始流动相中有机相的不同,二者在进样后由于其极性不一样,在同一波长下吸光度不一样而导致的“出峰”,一般通过改变测定波长可以掩蔽溶剂峰的出现与否。
当然在实际的分析方法开发中,很多人觉得溶剂的选择不是那么重要,往往忽视溶剂的重要性。下面是我做的一个项目中,由于溶剂选择不当,引发的一个惨案。
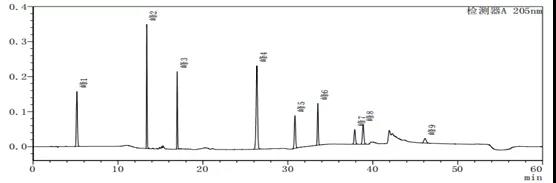
图7-1 系统适用性图谱
在峰4出峰位置出时常会有一个溶剂峰出现,导致在做定量限与检出限的时候,对结果造成很大影响。下面为不同的流动相A与流动相B的配比溶液作为溶剂,进样后空白溶剂图谱。
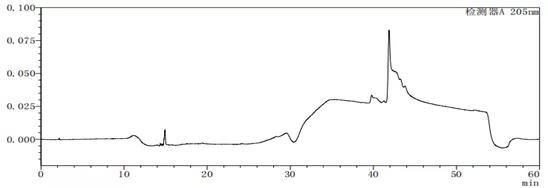
图7-2 流动相A:B=100:0的溶剂图谱
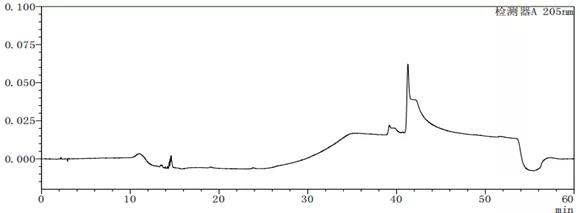
图7-3 流动相A:B=95:5的溶剂图谱
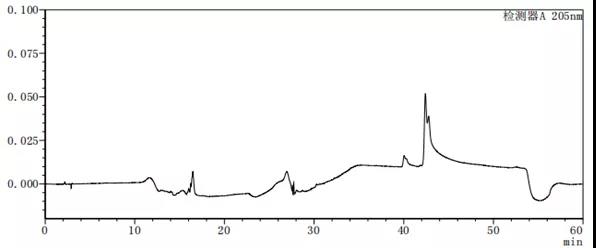
图7-4 流动相A:B=90:10的溶剂图谱
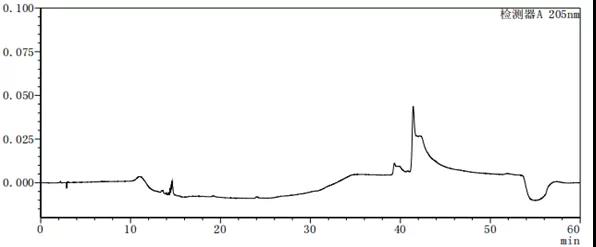
图7-5 流动相A:B=85:15的溶剂图谱
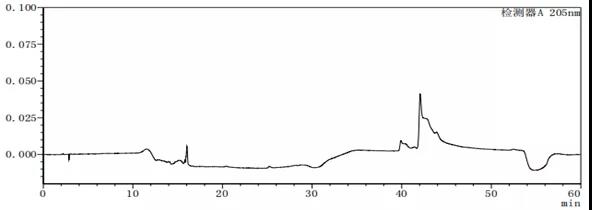
图7-6 流动相A:B=80:20的溶剂图谱
从上述图谱可以看出,不同的比例的流动相A与B混合出来的溶剂梯度峰是有差别的,在前期试验时,要尽可能多试几种不同浓度或种类溶剂,综合去考虑应该考虑哪种溶剂作为最终的溶剂。
路漫漫兮,其修远!吾将上下而求索。最后祝愿各位同行最想听到的一句话,实验顺利!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论