

































如下从催化剂的物料分类、金属催化剂、生物催化剂、其他信息4方面进行分析。
1.物料分类
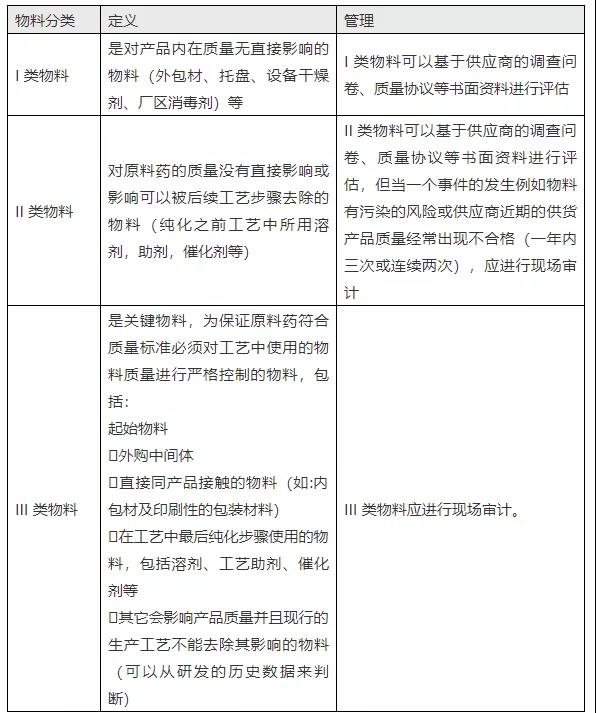
《2010 GMP原料药实施指南》指出,催化剂大多属于II、III类物料。涉及的信息如下:
“企业应基于对工艺的了解科学合理地定义哪些物料是关键物料(应在文件中体现),通常可以考虑将物料按照对于产品质量影响程度分为三个类别:I,II 和III类
2.金属催化剂
2.1金属试剂和金属催化剂中的金属残留情况分析(《2010 GMP原料药实施指南》)
在原辅料合成中可能用到金属催化剂或金属试剂,如铂、钯、锌、铁、铬等,这些金属可能在原料药中残留,它们可能以最初形式存在,也可能由于后续化学过程以其他形式存在。原辅料中残留的金属会进一步带入到制剂中。基于安全性和质量控制的要求,需要对这些残留的金属进行严格控制,并体现在质量标准中。
需要说明的是,本处提到的金属残留限度控制不适用于某些本身就含有金属成分的原料药,如:氯化钠中的钠含量不应作为残留金属控制范畴。
2.2 检测(《2010 GMP原料药实施指南》)
(1)关于分析方法
需要采用合适的、经过验证的、有一定专属性的测定分析方法。需要注意金属残留的形式可能不同于金属催化剂和试剂的初始形式。可以使用公认的药典方法,也可以使用其他适宜的测定方法。如果仅有第2 类或第3 类金属,也可以采用非专属性的方法。
基于pH3.5 有色金属硫化物沉淀的半定量测定方法通常不适用于金属的定量测定,但在某些情况下的常规测试中可能适用,如使用标准加入法或与其他专属性的测试方法配合使用。
(2)批检验结果,测试频率和标准中删除金属测定项目的考虑。
如果确定或怀疑合成过程会导致金属残留,则应进行定量测定。
如果合成过程显示金属可能被去除,常规测试有可能被非常规测试代替。如果连续多批中试产品或连续商业规模生产批次的金属残留持续远远小于浓度限度,则可认为金属残留物被充分去除。即使这样,质量标准中依然应当保留。
2.3 控制
《2010 GMP原料药实施指南》指出:对无机杂质(如催化剂)的试验内容和认可标准的制订应根据生产工艺,在开发阶段就应研究。硫酸盐灰分/炽灼残渣的检验方法和认可标准可参照药典方法。其他无机杂质可用其他合适的方法来测定,如原子吸收光谱。
《化学药物杂质研究的技术指导原则》指出,无机杂质是指在原料药及制剂生产或传递过程中产生的杂质,这些杂质通常是已知的,主要包括:反应试剂、配位体、催化剂、重金属、其它残留的金属、无机盐、助滤剂、活性炭等。
通常情况下,不挥发性无机杂质采用炽灼残渣法进行检测。某些金属阳离子杂质笼统地用重金属限度检查法进行控制。如需对某种(些)特定金属离子或上述方法不能检测到的金属离子作限度要求,可采用专属性较强的原子吸收分光光度法或具有一定专属性的经典比色法。虽然重金属检查法可同时检测砷,但因其毒性大,且易带入产品中,故需采用灵敏度高、专属性强的砷盐检查法进行专项考察和控制。
总之,我们总结:关于如何或具体的控制限度要求,目前可依照炽灼残渣、重金属、ICHQ 3D综合考虑。
3、生物酶催化剂
3.1生物酶概述
生物酶:是由活细胞产生的具有催化作用的有机物,大部分为蛋白质,也有极少部分为RNA。
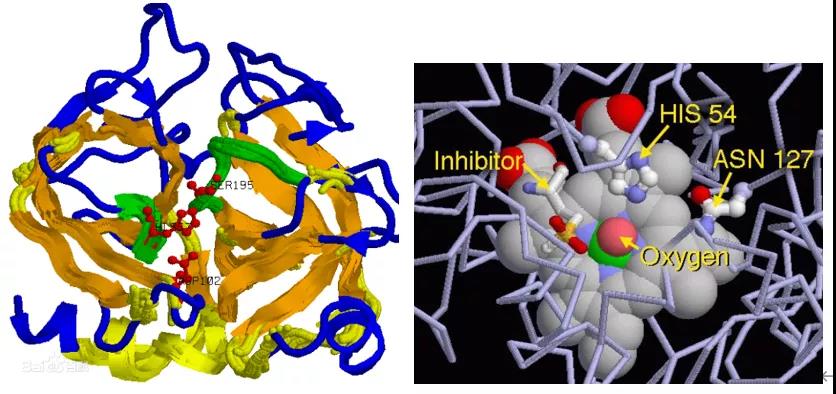
结构特性:生物酶是具有催化功能的蛋白质。像其他蛋白质一样,酶分子由氨基酸长链组成。其中一部分链成螺旋状,一部分成折叠的薄片结构,而这两部分由不折叠的氨基酸链连接起来,而使整个酶分子成为特定的三维结构。
生物酶是从生物体中产生的,它具有特殊的催化功能,其特性如下:

其与化学催化剂相比,特点为:

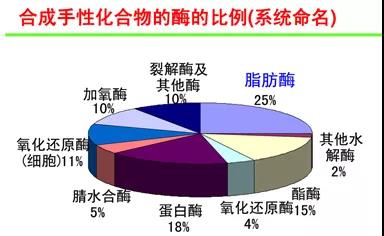
常用生物酶催化剂比例:
某一部或某些步骤采用了生物酶催化剂,该反应为生物合成和化学合成相结合。
实例:
如Lipozyme TL 100L 用于手性拆分普瑞巴林、用于催化合成蔗糖-6-乙酯(S-6-a)达芦那韦中间体的合成等;
Novozym435是一种应用广泛的脂肪酶,用于多种API,中间体的合成。
a,合成瑞格列奈中间体(±)3-甲基1-(2-(1-哌啶基)苯基)丁基胺
b,合成度洛西汀中间体2-氯-1-(2-噻吩)-乙醇
c,合成杀虫剂S—呋喃虫酰肼中间体 S—1-(2-甲氧基-3-甲基-4-氯-苯基)-2-丙醇
d,手性拆分(R,S)-扁桃酸乙酯
e,手性拆分阿伐他汀钙、罗素伐他汀、奥拉西坦中间体4-氯-3-羟基丁酸乙酯
f,合成非达司他中间体(2S)-6-氟-3,4-二氢-4-氧代-2H-1-苯并吡喃-2-羧酸
g,他汀类药物的重要合成原料3-TBDMSO戊二酸单甲酯
h,合成维生素E琥珀酸酯,维生素A棕榈酸酯,维生素A乳酸酯)
控制思路分析:
无法规参考;依据上文“关于金属试剂和金属催化剂中的金属残留”的控制思路总结为:
1、在原料药中残留,可能以最初形式存在,也可能由于后续化学过程以其他形式存在。基于安全性和质量控制的要求,需要对这些残留的生物酶进行严格控制,依据生物酶催化剂的种类建立合适的检测方法,以确定其在中间体或成品中的残留量。
关于如何或具体的控制限度要求,是药物研发和注册管理的范畴。考虑到大部分酶对人体无害且人体内也可能存在,在不影响制剂的生物活性前提下,可基于安全性的考虑,结合研发数据给予合理的控制限度。若采用了对人体有害的酶,则要求进行严格的控制并制定合理的限度。
3.2 生物酶催化剂质量标准的制定
对于制备API所用的催化剂,若在反应过程中无法将其去除或者参与了反应,对终产品的质量有一定的影响,因此需要对其进行控制,制定相应的内控标准。
《化学药物原料药制备和结构确证研究的技术指导原则》指出,一般内控标准应重点考虑以下几个方面:
(1)对名称、化学结构、理化性质要有清楚的描述;
(2)要有具体的来源,包括生产厂家和简单的制备工艺;
(3)提供证明其含量的数据,对所含杂质情况(包含有毒溶剂)进行定量或定性的描述;
(4)如果需要采用起始原料或试剂进行特殊反应,对其质量应有特别的要求,如对于必须在干燥条件下进行的反应,需要对起始原料或试剂中的水分含量进行严格的要求和控制;若起始原料为手性化合物,需要对对映异构体或非对映异构体的限度有一定的要求;
(5)对于不符合内控标准的起始原料或试剂,应对其精制方法进行研究,以利于对工艺和终产品的质量进行控制。
参考百度信息:生物酶的标准中,会存在酶活力检测项。
酶活力单位(U,active unit)1个酶活力单位是指在特定条件(25℃,其它为最适条件)下,在1分钟内能转化1微摩尔底物的酶量,或是转化底物中1微摩尔的有关基团的酶量。另外一个国际酶学会议规定的酶活力单位是Kat,规定为:在最适条件下,1秒钟能使1摩尔底物转化的酶量。
Kat和U的换算关系:1 Kat=6×107U, 1U=16.67n Kat
3.3 生物酶催化剂的质量协议相关的要求
动物/植物发酵还是提取,应根据供应商的生产工艺特点,进行审计;索取必要的声明,必要时应对以下情况在质量协议中作出规定:
- GMO(转基因),尤其是对植物来源和发酵法生产的物料;
- TSE/BSE(传染性海绵状脑病/牛海绵状脑病——疯牛病),尤其是对动物来源和发酵法生产的物料;
- 三聚氰胺/三聚氰酸、有机溶剂残留;
- 杀虫剂残留,尤其是对植物来源的物料;
- 毒素残留,如黄曲霉素、二氧(杂)芑等;
- 如果物料是用于无菌原料药生产的后续工序的话,还应当考虑对物料的微生物污染水平、细菌内毒素水平、异物等作出规定。
参考《2010 GMP原料药实施指南》
4、其他信息
4.1 CDE电子刊物相关信息
1. 《CTD申报资料中杂质研究的几个问题》(20121226):基于杂质谱分析的杂质研究是一种“以源为始”的主动思维模式,以“质量源于设计”的观点,从杂质来源入手,从制备工艺、化学结构、处方组成的分析出发,评估、预测产品中可能存在的及潜在的副产物、中间体、降解物以及试剂、催化剂残留等大体的杂质概况,辅以适当的强制降解、对照物质的加入等验证的手段,考证建立的分析方法是否能够将它们逐一检出,并进行相应的方法学验证工作;相比之下,传统的杂质研究是一种“以终为始”的被动行为和逆向思维模式,从杂质分析的结果出发,仅从建立的某种检测方法所检出的有关物质中归属其来源情况,而未充分分析与验证可能存在的潜在杂质情况,建立的分析方法能否全面检出这些杂质,故容易出现杂质漏检的情况,难以全面掌握产品的杂质谱。
2.《CTD第一期研讨班共性问题汇总及解答》(20110613)
十八、对于原料药,详细的杂质研究报告应放在哪个部分?
回答:杂质研究是原料药质量控制研究中的重要内容之一,主要研究信息包括杂质谱的分析(根据合成工艺对潜在的工艺杂质进行分析、根据降解研究对可能的降解产物进行分析)、分析方法的建立和验证、杂质限度的确定等。在CTD格式文件中,3.2.S.3.2重点是结合生产工艺和降解途径研究对产品中潜在的杂质(包括有机杂质、无机杂质、残留溶剂、催化剂)进行分析并说明控制情况;3.2.S 4.3中需要提供杂质分析方法的建立和验证信息;3.2.S.4.5中重点是阐述杂质控制限度的制订依据,包括对比研究的结果。
3.《加拿大对合成用起始原料的相关要求简介》(20070706):为了评估合成用起始原料中所有潜在的杂质,包括几何异构体、光学异构体杂质、毒性杂质、残留溶剂和催化剂等,应提供该起始原料的合成工艺简介,包括从简单的化合物分子开始,到该起始原料的整个合成工艺概述、所有用到的试剂、溶剂和特定的中间体、合成路线图。应详细说明在该起始原料制备过程中可能残存的病毒、细菌、残留的蛋白质和疯牛病毒等危险物质,并对其残留的可能性进行分析。
4.限度
Organic Process Research & Development中文献报道[1],常规口服固体制剂中蛋白残留,建议参照ICH指导原则中非特定单杂限度控制,控制限度为0.1%,在物料C-3中拟定酶蛋白残留限度不得过0.1%。
[1] OrganicProcess Research & Development. Andys wells etc. The use of enzymes in themanufacture of API’s – a science and safety-based approach to ensure patient safetyand drug quality.
5.总结
综上,制备API的过程中引入的生物酶催化剂,作为工艺杂质,需要进行控制。但国内外尚无明确指南提及生物酶催化剂的控制思路。可参考杂质控制的一般原则进行控制。



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论