


































1 序言
在药物发现的早期,药物化学家更多地专注于分子的PK性质、有效性和毒性。随着药物发现技术进步和新药开发流程的优化,越来越多的研究者认识到在药物发现阶段即为分子构建良好物理化学性质,对下游的开发阶段有重要的意义,从根源上解决可能存在的生物利用度、稳定性或可生产性问题,无疑会降低下游开发的难度,提高开发效率。此外,伴随高通量筛选技术的发展,大量备选化合物得以产生,有效性和毒性也不再是唯一的选择标准。
另一方面,新分子实体的晶型/盐型筛选获取的大量固体型式,也为后期的药物开发提供了更多的选择。化合物及其晶型/盐型开发为药物的难度和代价往往成为分子取舍的重要内容,即所谓的可开发性。而化合物的稳定性往往是最为重要的筛选指标之一。在药物发现之后的开发阶段,合成工艺的放大、非临床动物制剂的处方确定以及人用临床制剂的处方工艺开发等工作,也与化合物的稳定性息息相关。
2 稳定性的判断标准
在药物研发的进程中,决策总是伴随资源、风险和收益的平衡。对化合物的稳定性是否满足进一步开发的条件的评判也是决策平衡的结果。
不同化合物或不同固体型式之间、不同处方之间的稳定性高低比较是容易实现的,但是稳定性是否可以接受往往是难以界定的。对于一般上市的制剂而言,有效期的规定多在24个月。同样地,一般认为API应当满足的常规条件下24个月的货架期(最好是室温下)。API和制剂是否满足货架期,进一步取决于质量标准中有关物质的限定。关于含量,ICH原则有明确的规定,含量降低超过5%是显著的变化,可以考虑将此限度作为最低接受限度。而对于单个杂质的增长而言,却没有特定的限度。
对于新药,可以通过专门的非临床毒理试验对杂质水平进行界定,从而在研发过程中放宽杂质的限度,但是代价可能是时间和金钱。ICH原则中确定了界定和鉴定限度,而这也与临床用法(最大日剂量)有关,在研究早期,这些信息往往是极为模糊的。因此,API和制剂中出现的杂质都可能面临结构鉴定或者杂质获取,这项工作的难度将会影响后续的开发工作。此外,对于疑似基因毒性杂质也应当引起足够的重视,其极低限度可能为后期研发带来巨大的麻烦。
化合物的稳定性是否可接受,也跟公司的决策有重要的关联。化合物或者制剂,无论是在生产还是在储存过程中体现出不稳定性,理论上讲,都是存在解决手段的(除去极少特别极端的例子)。但是化合物越不稳定性,后期开发和商业化的代价也就越高。这种代价是否值得,是与其临床价值和其商业收益密切相关的。
此外,是否存在可用的解决手段,也影响化合物的稳定性接受程度。例如掌握“活性层包衣”这一生产技术的公司,在处理极不稳定的化合物的口服制剂开发时,会显得游刃有余。而采用独有技术开发的药物,被仿制的难度越大,盈利期可能更长。化合物的稳定性对后期的药物开发风险有重要影响,其判定标准受到众多因素影响。
笔者认为伴随项目立项,就应当明确开发目标,并且预设开发路径。根据其中与稳定性相关的指标可能包括剂型、可接受的储存/生产条件、有效期等。有针对性的对药物发现阶段的化合物有针对性的进行评价,可以使药物发现中化合物及其固体型式的优化和选择的决策能够高效而准确的进行。
3 方法学
(1)理论分析
从结构入手分析化合物的稳定性是最基本的手段。可以获得对稳定性有初步的认识,虽然无法获取稳定性的完整结果,却可以指导后续的稳定性试验设计和数据解读。此外,将化合物与结构类似的化合物对比分析,也有助于对化合物稳定性的认知。
(2)分析方法
良好的分析方法,是稳定性研究的基础。对于早期发现阶段而言,分析方法的开发宜追求适度原则。例如不同化合物之间的比较评级,分析方法可以适当粗略,可以区分稳定性即可。对具体化合物的评价而言,可能涉及具体杂质增长的分析,方法应当适当优化,以区分和定位主要杂质,甚至有时为了明确某些问题,采用质谱解析或者杂质定位等分析手段也是必要的。
在分析方法开发过程中,强制降解试验是获取杂质谱和优化分析方法的常有手段。即将化合物置于各种强制条件(强酸、强碱、热或者光),关注杂质的产生情况。参考文献1中对各种可以选择的降解条件有较为详尽的叙述(也可参考药事纵横占小兵、梅希的文章《强制降解试验怎么做?稳定性指示方法怎么建?别急,大神给出了答案》),本文不再详述。
对于分析方法开发来说,分析研究者更多地关注强制降解杂质结果。虽然强制降解的过程并非药品最终面临的环境,过程中产生的很多杂质在最终的药物中可能不会产生,但是强降解试验对化合物的稳定性和杂质产生机理仍有较好地指示作用。对制剂研究者而言,对化合物的稳定性研究更多地应当关注杂质产生的机理,以预测制剂开发过程中可能出现的稳定性问题,以及可能的应对手段,以供关键节点的决策。因此,制剂研究者在强制降解的数据解读的过程中,需要特别注意将强制降解过程产生的杂质与降解条件联系起来。
(3)稳定性研究
候选化合物的稳定性研究一般包括API生产过程中的稳定性和储存稳定性,在药物发现阶段最容易被忽略的是与制剂相关(无论是人用临床制剂,还是非临床评价用制剂)的稳定性研究,研究内容一般包括生产、储存和使用三个方面。
生产过程持续相对较短,其稳定性预测往往比较简单。但这需要研究者基于对工艺过程的深入理解,才能设计出合理的考察方案。无论是化合物本身,还是制剂的生产,主要是由单元操作组成。结合强降解的结果,合理地选择单元操作进行试验即可评价生产上的稳定性是否可行。但是需要注意的进行边界挑战,因为在实际的生产过程中,比小规模的试验的时间更长,条件波动性范围更宽。考虑相应的生产设备因素和药品包装也是必要的,尤其是对于液体制剂。此外,在关注化合物化学稳定性的同时,物理稳定性的结果也需要特别注意。
粗略地研究候选化合物与常见辅料的相容性也是必要的。需要注意的一点是,早期的研究目的是更多地暴露问题,因此可以适当选择生产过程的最劣条件进行考察。例如,在对口服固体制剂的候选化合物研究时,采用粉碎后的化合物,采用压片的型式代替简单的粉末混合等。在很多情况下也不需要获取精细的结果,因此采取一些高通量的表征技术可以极大地提高试验效率,如等温微量量热法等。
(4)货架期预测
在稳定性研究中,最具有挑战性的内容便是货架期的预测,尤其是固体制剂的货架期预测,相关的研究法方法也在不断的演进中。而这些方法的理论基础都是化学反应动力学,即以Arrhenius方程为基础,通过提高温度增加反应速率,从而对常规储存条件下的降解情况进行预测。
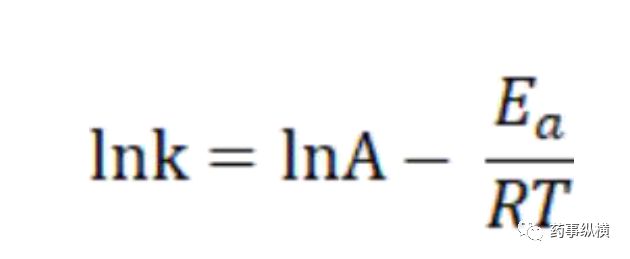
按照上述方程,反应速率的与绝对温度的倒数成正比。
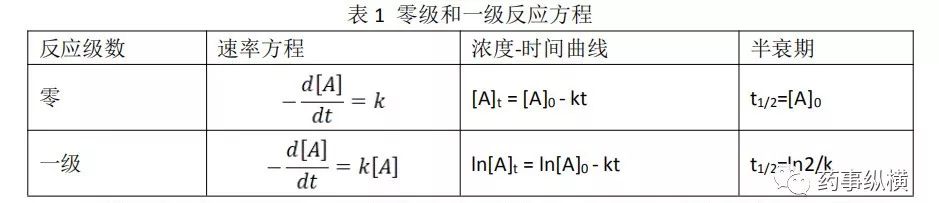
一般认为,在药物中的化学反应,尤其是在降解反应的早期,从表观反应速率上看,大多数符合零级反应或一级反应,即所谓的假零级反应或假一级反应,相关的速率方程如表1所示。
将样品置于不同温度下,在特定的时间点取出样品,分析样品中化合物的含量或效能。即可计算不同温度下的反应速率,从而通过Arrhenius方程计算非试验温度下的反应速率,再通过反应速率方程,预测相应的含量或效能。
该方法的预测的准确性受到多重因素的影响。分析方法正确且准确,环境温度湿度等条件的精准控制是基本条件。对研究者而言,更多的精力需要花费在试验设计上。
一个基本的试验设计一般采用四个取样时间点,四个温度条件。四个浓度-时间数据可以较为准确地拟合选择反应方程,进而确定反应级数。在反应时间的间隔的设置上,需要考虑转化率,有研究者指出,降解程度为15~20%时,计算反应速率最为准确。但是在早期研究中,对反应的进程往往知之甚少,一般按照等时间间隔或者按照指数增加的方式设置取样时间,也可以根据已有结果灵活调整取样时间。
此外,值得一提的是,不同温度下的反应速率不同,因此,采用不同的取样时间,有利于获取更为准确的结果。在早期的研究中,获取精确的反应速率数值没有必要,直接假定反应级数,选择1到2个时间点获取反应速率用于后续的计算评估也未尝不可。
对于温度的设置最主要的考虑是Arrhenius方程的适用性。引发反应速率和温度关系偏离该方程的主要原因主要包括温度变化引起的反应机理的变化、相转变(Tg、Tm、去溶剂化等)、pH变化、限速步骤变化、湿气影响等。例如,对于水解反应,lnK与1/T的关系呈现碗型,一般意味着反应机理的变化,钟型意味着限速步骤的变化。
在考察温度的选择之前,需要通过DSC等手段对化合物的相变有一定了解,若是水溶液考察则需要考虑合适的缓冲体系,同时固体进行考察时需要考虑对湿气的控制等。选择四个温度点,通过线性拟合即可较好地评判所选体系是否符合Arrhenius方程,同时建议将所需要预测的温度包含在内,以便对预测结果不断进行验证。在不需要非常精确的计算结果的情况下,选择三个温度甚至两个温度也是可以的。
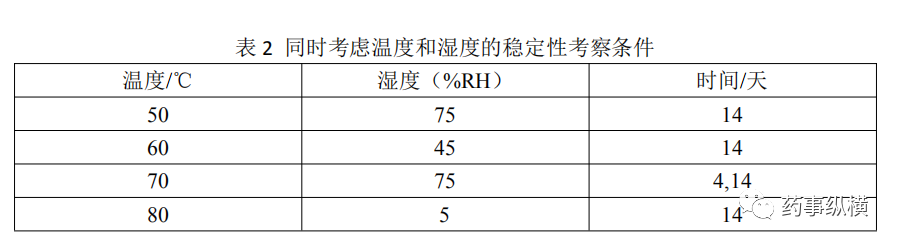
有研究者将温度和湿度结合,同时为稳定性考察的加速条件,构建起所谓的等转化方法,并推荐了一些适合在原辅料相容性考察应用的条件(见表2)。这种更为简单快捷的方法更加适合早期研究过程中的化合物取舍或原辅料相容性试验。
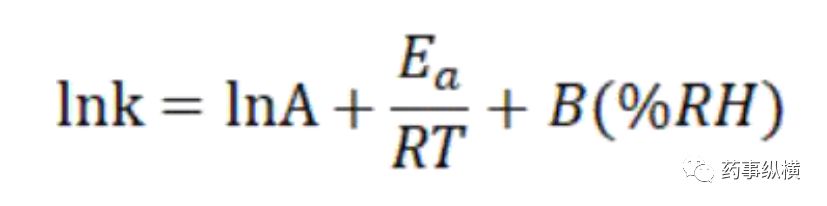
4 总结
稳定性研究是一个系统的工作,其最终目标在于保障上市制剂的稳定性。而对于新药开发这场马拉松而言,稳定性研究的不同阶段有不同的内涵,不同的研究者有不同的关注内容,因此,知识的不断积累和团队的沟通合作极为必要。虽然法规一般要求在临床III期前完成全面的稳定性研究,但是在化合物进行临床前或进入其他关键节点前适当地进行稳定性研究是十分必要的。相对于稳定性在开发后期可能引发的问题和风险,早期稳定性研究的微弱投入在后期都将获得巨大回报。
在实际研究的方法学选择上,稳定性研究也不仅仅只是考虑法规的要求,更多地需要从对化合物性质构建全面科学理解的需求出发。这种基于科学本质的理解,能够更为有效地支撑药物制剂的后续稳定开发,降低过程的反复和风险,提高药物研发的效率。
参考文献:
(1)Blessy M et al. Development of forced degradation and stability indicating studies of drugs-A review. Journal of Pharmaceutical Analysis 2014; 4(3): 159-165;
(2)Developing Solid Oral Dosage forms: Pharmaceutical theory and practice, charter 5, Drug Stability and Degradation Studies;
(3)Kenneth C. et al. Accelerated aging: Prediction of chemical stability of pharmaceuticals. International Journal of Pharmaceutics 293(2005)101-125;
(4)Kenneth C. et al. Hydrolysis in Pharmaceutical Formulation. Pharmaceutical Development and Technology, 7(2), 113–146 (2002).



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论