
































脊髓性肌萎缩症
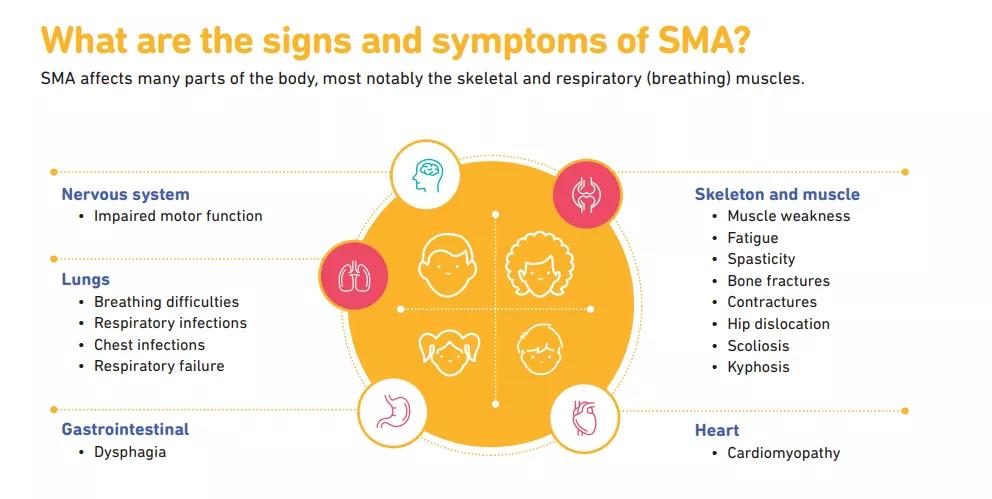
脊髓性肌萎缩(Spinal muscular atrophy, SMA)是一种罕见的常染色体隐性遗传性神经肌肉疾病,位于2岁以下儿童致死性遗传病的首位;主要发病原因是由于患者SMN1基因的缺失或突变,导致全身功能性SMN蛋白表达不足。该疾病以脊髓和下脑干中运动神经元的丢失为特征,从而导致严重的、进行性肌肉萎缩和无力。最终,SMA患者可能丧失行走能力,并且难以完成呼吸和吞咽等基本生活功能,从而导致重大的医疗干预和护理帮助。如果不进行治疗,大多数患有严重疾病类型的婴儿在没有呼吸干预的情况下,无法活到两岁。
2018年5月SMA被纳入国家《第一批罕见病目录》。2019年《脊髓性肌萎缩症多学科管理专家共识》推出,2020年《脊髓性肌萎缩症遗传学诊断专家共识》推出。目前中国国内获批的SMA相关新药有:诺西那生钠注射液(英文商品名Spinraza®)、利司扑兰口服溶液用散(英文商品名Evrysdi®)。
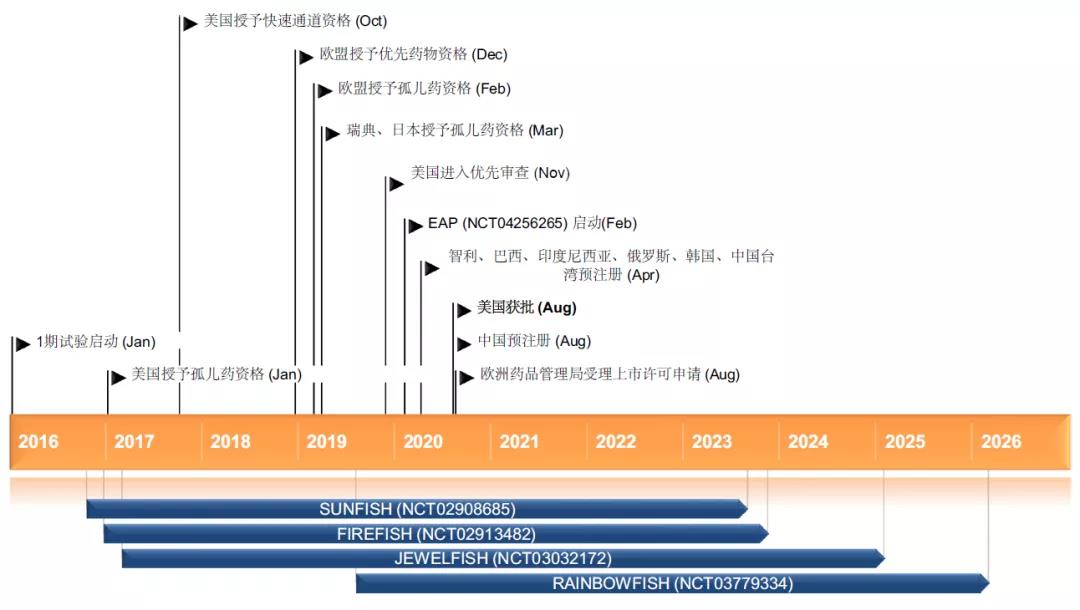
2021年6月,中国国家药品监督管理局(NMPA)正式批准了罗氏利司扑兰口服溶液用散(中文商品名:艾满欣®,英文商品名:Evrysdi®),用于治疗2月龄及以上患者的脊髓性肌萎缩症(SMA)。研究结果显示,利司扑兰治疗后的1型SMA患者生存率较之自然史显著提高,实现运动里程碑,呼吸和吞咽功能获得改善。对于2型和3型SMA患者,用药后运动功能及生活独立性获得改善。没有任何与药物相关的安全性事件导致退出研究。

2021年8月中旬,罗氏在中国市场正式推出本品。
药融云数据(www.pharnexcloud.com)显示,60mg/瓶的零售价为6.38万元;此外公司也推出了慈善援助计划。

利司扑兰口服溶液直接靶向疾病的潜在分子缺陷,增加中枢组织和外周组织的功能性SMN蛋白的产生,该药由罗氏旗下基因泰克(Genentech)与 SMA 基金会和 PTC Therapeutics 合作研发。
2020年8月发布的《中国SMA患者生存现状白皮书》报告显示,对于病情最为严重的1型SMA患者,2016 年之前出生的1型SMA患者,从发病开始,人均需要 6.6 个月才能获得确诊;而2016 年之后出生的1型SMA患者,其确诊时间需要3.5个月。多位医学专家呼吁:在用药可及性大幅提升的情况下,SMA的早诊早治、多学科管理和康复训练,已成为延缓疾病进展的关键。
每年的8月7日为“国际脊肌萎缩症关爱日”;药融圈一直关注孤儿药领域发展,呼吁更多医药企业及专业医药投资基金关注各类罕见病的药物研发。



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论