


































新药开发以普通口服固体制剂居多,制剂经过胃肠道吸收进入体循环,早期制剂以获得足够的体内暴露为目标,以便研究新的化合物分子在体内的药理学,药代动力学和毒理学情况。根据这几年从事新药开发的状况来看,制剂人员更多是被动接受新药药物分子结构以及确定的固态开发形式,在前期药物发现阶段,参与的深度还是较低的,当然每个公司的开发策略与模式是不同,而且其中还涉及项目的保密。但是,万变不离其宗,拿到一个新的化合物,还是要从性质入手,包括其结构特征,理化性质,生物学性质以及PK,毒性等。在新药开发早期,API的可及性往往限制研发的推进,如何在化合物众多理化性质中把握关键性质,抓住主要矛盾,将是我们制剂开发人员需要思考的问题。
生物药剂学分类系统(BCS)的横空出世,似乎给了我们指引关于新药早期开发需要格外关注的药物性质-溶解度和渗透性,其背后的关键性质不仅是获得体内生物等效性研究豁免的有用工具,还是FDA建议的新药发现和早期开发决策的有用工具。当然,除了BCS所要关注的药物性质之外,化合物的稳定性在药品开发中也是需要足够关注。预计通过几篇小的文章,给大家介绍一下,新药早期开发中需要关注的关键的药物性质,包括溶解度,渗透性以及稳定性。本文主要介绍一下有关药物溶解度的相关信息。
历史上,本人在药事纵横也是谈论过溶解度的,如果大家感兴趣也可以去去回顾一下:
对于溶解度的认识确实是不断的发展与深入,而且不同的书籍与著作可能采用不同的观点与方向去讲诉有关溶解度的故事,随着学习的加深,确实也获益匪浅,一定程度上,避免了一直被领导支配的恐怖,他可能上嘴唇碰着下嘴唇,下面的人就要跑断腿。说的有点夸张,确实也看到有的同事反复去执行领导的要求,也做了很多次平衡溶解度。对于固体制剂开发怎么重视都不为过,但是,是否需要反复去确认呢?
以下几个问题供大家思考:
- 是否可以通过其他的性质间接去证明溶解度未发生变化?
- 多晶型影响溶解度吗?
- 粒径及粒度分布改变了是否需要重新研究平衡溶解度?
- 非pH依赖的药物分子如何设计各介质条件?
- 怎么确定平衡溶解度达到平衡,还是所有化合物24h即可?
以上问题或许无法在本文找到答案,可以看看本人历史上的小文,或者借助工具书,去探索究竟。下面我将开始正式开始本文的进程(以下提高的溶解度概念皆指平衡溶解度,除非特别说明)。
基本概念:溶解系指一种或一种以上物质以分子或离子状态分散在另一种物质中形成均匀分散体系的过程,溶液中任意部分都具有完全一致的性质。在溶解的物质(即溶质)分子与分散介质(即溶剂)分子产生相互作用时,如果不同种分子间的相互作用力大于同种分子间作用力,则溶质分子从溶质上脱离,继而发生扩散,最终在溶剂中达到平衡状态,即溶质的溶解速度与其结晶速度相等。所以也可以说,物质的溶解是溶质和溶剂的分子或离子相互作用的过程,这种相互作用力主要是范德华力、氢键力和偶极力。
以上说法,其实很多书和我以前写的小文都提到过,下面的图1不少文章也有,再次去介绍关于平衡溶解度的概念,其实还是想重申平衡的含义:溶质脱离晶体结构中的溶解速率与溶剂中溶质的析出达到了动态的平衡。这样就要求我们,一来我们做平衡溶解度试验的时候需要根据化合物的大概溶解度情况(预实验或者计算的方式),抑或着加入大量API于介质中(新药早期开始对于溶解度比较好的化合物还是需要不少量的),保证平衡时溶质仍存在未溶解的部分。二来对于什么时间达到平衡,不同的化合物且相同的化合物不同的介质环境应该都是有所差异。有的化合物24h,48h甚至72h。如何证明24h达到平衡,起码你需要再检测24h以后的一个时间点,证明后一个时间点与24h溶解度无明显差异。至于如何说明这个无明显差异,这个尺度如何把控,暂未发现报道。三来平衡是溶液所具有的浓度为该化合物的饱和溶解度。无论化合物成盐,转变为无定形或者亚稳定性态,其平衡溶解度应该都是一致的。
有必须说明一下,不同固态形式可能可以提高溶解度,但是也是有条件要求的。比如化合物成盐提高溶出速率,形成过饱和溶液,看似溶解度提高了,可是如果在特定pH介质且缓冲能力足够强,就可以消除成盐反离子的作用,化合物无论成盐,还是游离碱,游离酸,溶解度是相同的;对于化合物无定形来说,看似也是提高了溶解度,无定形固体分散体以极小的粒度,小到可以达到分子级别,而且无定形的API需要亲水性载体来稳定,这样也提高ASD的润湿性,而且无定形本身就是打破了溶解所需要的晶格的限制,破除了高熔点固态化合物溶解的限速步骤,这样提高最终制备的ASD中药物的溶解度似乎板上钉钉。但是,首先需要明确一定,ASD其实是不具有平衡溶解度,因为无定形态固有的结晶趋势,因此实际测量的溶解度可能无法达到最大可达到的无定形平衡溶解度。为了确定无定形态到底能提高把API的溶解度提高到了多少,可以通过理论公式去计算。
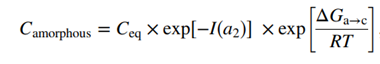
ASD开发化合物的溶解度理论上增强可以是几个数量级,大多数药物溶解度可以提高2-20倍,对于极高熔点的,甚至可以提高50倍。然而,当将几种无定形药物的预测溶解度值与文献中的实验报告值进行比较时,通常存在较差的相关性。这种不一致部分归因于无定形态物质快速结晶成亚稳定性态,接着又会由亚稳态向稳定态转变,最终溶液浓度和稳定型晶型的平衡溶解度一致。通过上面的解释,这样我们就可以理解,物理药剂学上对于溶解度的定义:药物在溶剂中的热力学溶解度是指一定温度和压力的平衡条件下,在一定体积的溶液中所含有的最稳定晶型药物的最大值。热力学溶解度一般指平衡溶解度。
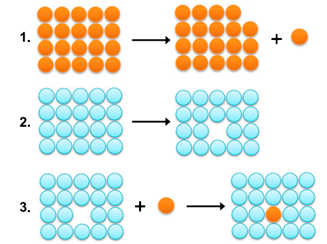
图1.溶质药物分子从固体颗粒中置换出来并进入溶液需要三个基本步骤。第一步:从晶格中去除单个溶质分子;在此步骤中需要能量来克服固态中的溶质-溶质相互作用。第2步:在溶剂中形成空隙以容纳溶质分子。尽管此步骤也需要能量,但它可能比步骤1所需的能量低得多。步骤3:溶质分子插入溶剂中,形成溶质-溶剂相互作用。简单地说,如果溶质-溶剂相互作用(即第3步)释放的能量大于第1步和第2步所需的能量,则对于溶解度是有利的。
溶解度的限速步骤:由图1可知,药物溶解度有两个主要决定因素:1)克服溶质固态分子间力强度所需的能量和2)能量溶液中溶质和溶剂分子相互作用产生的(溶剂化)。
根据一般溶解度方程(GSE),可以估算药物在水中的固有溶解度(S0):
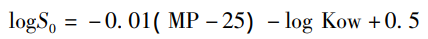
MP为熔点(℃),Kow为油水分配系数。其实,这个方程,也介绍不少次了,该方程很好的契合了图1中所描述的一个晶体药物的溶解过程,提取了两个重要的可能限制溶解度的参数,即晶型药物的熔点和logP。当然,同一化合物不同的晶型之间熔点是不同,其可以表征固态形式的差异,是反映化合物固态形式的固有属性的一个参数。LogP反应化合物亲脂性的参数,更加强调分子结构的特性,与固态形式无关,即同一分子LogP是相同的。这样也很好解释上述提出的一个问题:多晶型影响溶解度吗?
多晶型之间熔点的差异,LogP一致,带入上述公式,是不是表明其溶解度发生变化了。那么问题来了,合成可能合成很多批次的原料,晶型不变,还需要反复去验证溶解度是否发生变化吗?
其实,从上面的计算公式,我们还可以推出新的结论。当溶解度数值发生1个对数单位变化,LogP也会相应的变化一个对数单位。而熔点需要变化100℃的情况下,溶解度才发生1个对数单位变化,可见LogP对于溶解度的影响可能远远大于熔点。这样也解释了一般认为200多℃为高熔点,但是仍有化合物溶解度可以达到200μg/ml以上的溶解度;而熔点100多℃,仍旧可能是难溶性药物。
最后还要补充一点的是,有的文献/书籍中说化合物的溶解度是其化合物的固有属性,固有属性可以理解为在条件一定下,其溶解度应该是确定不变的。可是,有的资料又说,化合物溶解度并没有单一的数值。一开始感觉这两份资料,是不是有点矛盾。其实,在药典里面,很少去报道该化合物具体的溶解度数值,因为化合物的溶解度的数值的测定具有不确定性,影响因素太多了,包括化合物分子结构本身的属性,固态形式,固态形式纯净程度,配制的介质,试验操作(离心VS过滤膜,离心多久,会不会因为离心过程温度变化结晶析出)等等。从理论上说,控制这些变量,理论上在一定条件下,溶解度是确定的。可是在真实的世界,平衡溶解度的数值又很难去统一。当然,即使有些条件无法全部统一可能造成结果有偏差,但不可能具有质的变异,除非你的试验出现了重大的失误,设计具有问题。
小结:新药的开发,项目是无比的急切地,可能在实际研发中多次变更整个IND申报的试验计划,轮到制剂这边往往,计划可以推迟,但是计划的终点依旧不变。如何在所剩不多的时间中,掌握主动权,即使痛,还要快乐着(强颜欢笑),那就需要我们不断地武装自己,在平时夯实基本功,提高对制剂开发中的相关知识掌握的深度,形成自我的知识体系。
最后絮叨一句。以上除了一些书本和文献中的内容,还加入了不少的个人的理解与感悟,如有不妥,还请及时反馈,以免给予大家以误导!
参考文献:
1.Strategies to Address Low Drug Solubility in Discovery and Development
2.Martin物理药剂学与药学。(书籍)
3. Amorphous solid dispersions: Utilization and challenges in preclinical drug development within AstraZeneca
4. Pharmaceutical Amorphous Solid Dispersions (书籍)
<END>




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论