


































许多先导化合物尽管有效,但由于其溶解性不足导致生物利用度较低,因此未能进入临床试验,这使得溶解性成为早期药物发现中的关键因素。在不损失药效的情况下改善难溶的先导化合物的溶解性是药物化学家在药物发现过程中面临的一项具有挑战性的任务。溶解度不仅是分散或液化一种物质的重要因素,而且也是达到理想治疗效果所需的体循环中最佳药物浓度的重要因素。据估计,新开发的分子中有40%以上实际上不溶于水。溶解性差的分子不仅给体外和体内分析带来困难,而且增加了药物开发的负担,表现为优化溶解度所需的时间更长,成本更高。
世界各地的研究人员都在努力开发新的方法和策略,以在不影响生物活性的情况下提高先导化合物的溶解度,一些物理方法方法包括减小颗粒尺寸、药物在载体中的分散和络合、使用缓冲液、盐析、共结晶、微粉化、纳米化等[1]。其中,以结构修饰为核心的药物化学方法是一种通用而独特的方法,在改善溶解度的同时,它还可以同时改善其他药代动力学或物理化学参数。
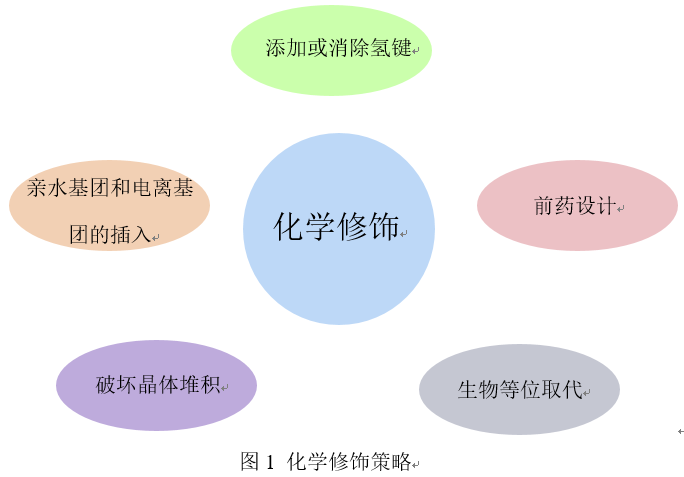
在这篇文章中,首先简要介绍了溶解度及其影响因素,然后列举了最近文献中报道的不同结构修饰策略的成功例子,包括亲水性和电离基团的插入、添加或去除氢键、前药的合成、分子对称性和平面性的破坏、生物电子等排体替代等[2]。
一、药物水溶性的本质
溶解的热力学过程主要包含两个部分:过程一是物质晶体在溶液中解离成单个分子,过程二是单个分子被水分子包裹形成水合物。从能量过程来看,两个过程分别对应着晶格能和水合能。
ΔG(溶解)=ΔG(水合)-ΔG(晶格)
故减小晶格能,增加水合能,即可促进溶解,晶格能与化合物本身的结构、极性等有关,分子之间作用力越大,晶格能越高,化合物表现出较高的熔点和较差的溶解性;水合能由药物与水分子的作用方式所决定,与化合物本身的结构、极性和离子化性质相关。
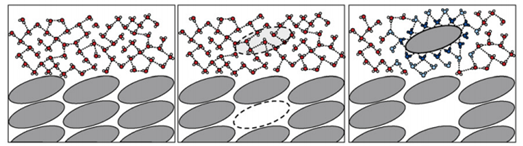
图2 溶质热力学溶解过程[3]
二、水溶性的影响因素
能够影响药物水溶性的理化参数有LogP、分子量、熔点、PSA (极性表面积) 和 pKa等。Jain和Yalkowsky溶解度计算公式:
LogS=0.5-LogP-0.01(MP-25)
由此公式可以看出化合物LogP值和熔点对溶解度S影响,根据经典的“Lipinski 五规则”,药物分子的LogP值应不大于5,PSA(polar surface area,极性表面积)指分子中极性原子表面积的总和[4]。根据Veber规则,药物分子的PSA要小于140Å2,最适宜的PSA范围一般为50~80Å2。pKa显示的是化合物的离子化能力,现今上市的药物大部分都具有弱酸性或弱碱性,这使得化合物可被一定程度的离子化,易于溶解。
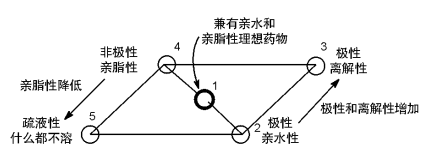
图3 化合物亲脂性和亲水性的的关系[4]
三、通过化学修饰改善药物水溶性策略
3.1插入亲水基团和可电离基团
添加亲水基团和可电离基团是改善溶解性的最常见和最经典的策略。据报道,亲水性基团和可电离基团的插入可以通过降低先导化合物的LogP值提高溶解性。另外引入亲水基团实际上是增加化合物的水合作用,促进溶解的热力学过程,可电离基团离子化过程会提供额外的能量,促进溶解。
可供选择的亲水基团一般为直链或环状的含有N、O原子的基团,如直链的胺类、醇类,环状的哌嗪、吗啉、氧杂或氮杂环烷烃、酸碱等可离子化片段等。值得注意的是,这个策略成功的关键在于引入恰当的亲水基团而不影响化合物的其他重要指标,如活性和安全性。
- 案例1:
Rasina和他的同事们致力于开发纤溶酶抑制剂,并发现了最初的化合物1的溶解性很差,该团队通过从四氢呋喃中去除苯基和在5位插入可电离基团实现了溶解度的改善。化合物2-5具有较高的热力学溶解度,较低的脂溶性,并且与母体化合物1相比,纤溶酶抑制活性显著提高了。
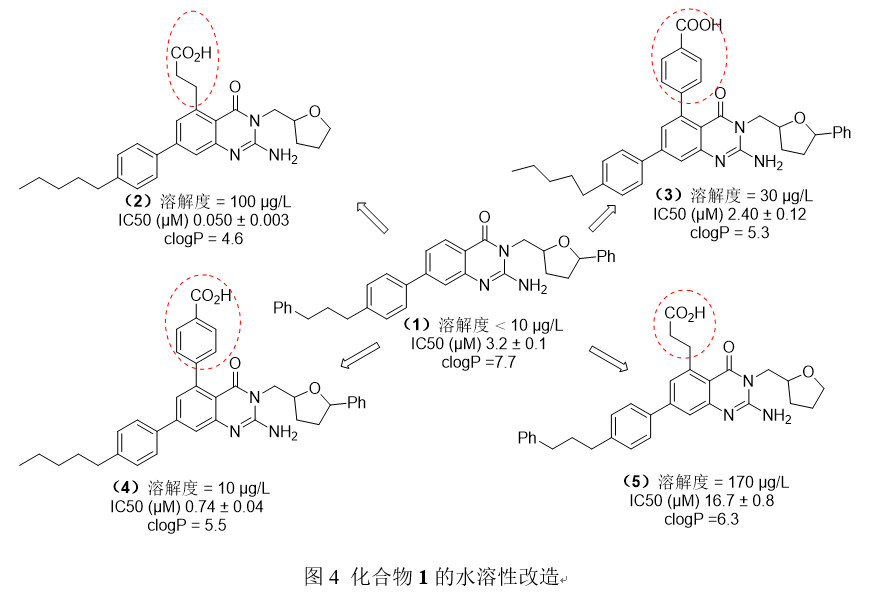
- 案例2:
抗疟疾药物青蒿素6,在水中的溶解性较差,为了提高其溶解性,合成了具有较高溶解度但不稳定的羧酸类似物7的钠盐。Li Ying和同事们制作了青蒿素的胺类衍生物8和9,它们具有更好的热力学水溶性和稳定性。
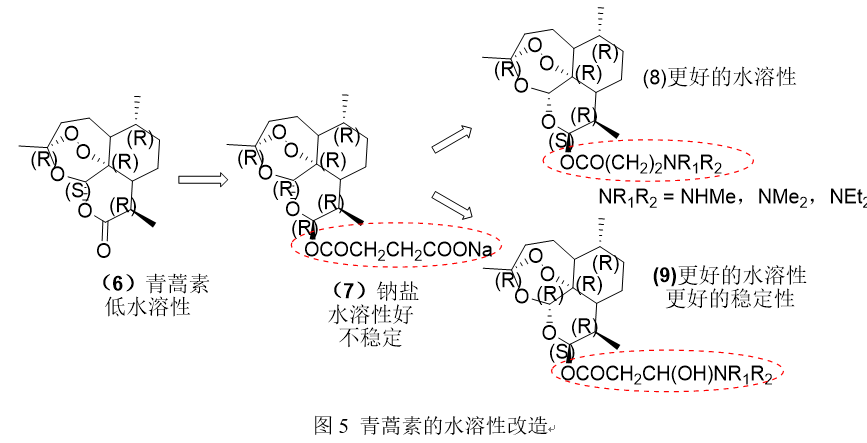
- 案例3:
Rilpivirine(10)是一个非核苷类逆转录酶抑制剂,对野生型HIV-1,和两种主要的突变体均有较好的活性,但水溶性仅有0.02µg·mL−1。在含氮母环的6位引入吗啉环后,化合物的溶解度得到了较大的提高。化合物10d在野生型和突变体的活性得到了较好的保持,其溶解度提高到14.2µg·mL−1,提高了700余倍。
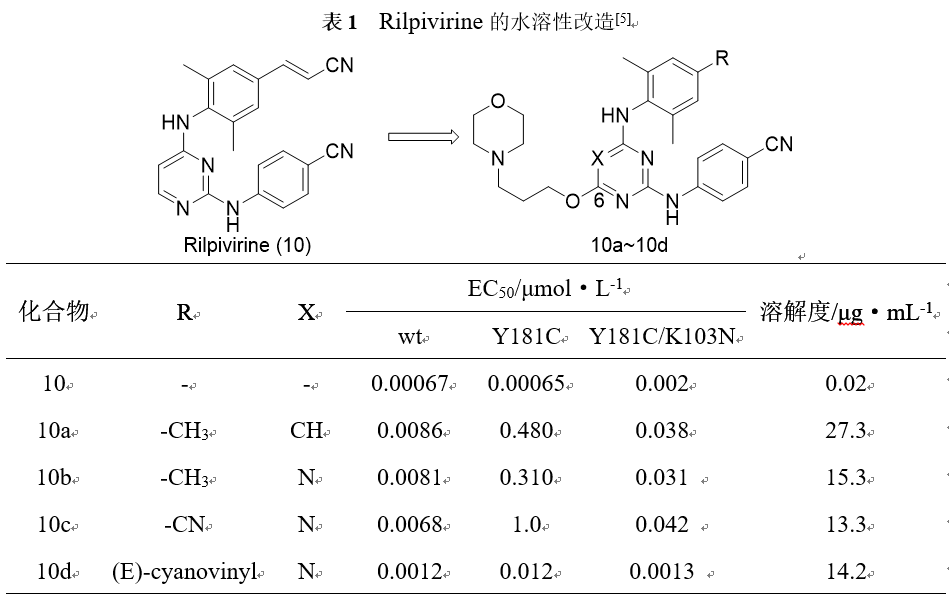
3.2添加或去除氢键以增加溶解度
氢键供体和氢键受体如羟基和氨基基团的安装,可以通过与水分子形成氢键来提高水的溶解度,也有报道说,去除氢键可能通过降低熔点而导致溶解度的增加,使其成为一种有效的改善溶解度的方法。3.2添加或去除氢键以增加溶解度
- 案例1:
沙利度胺11由于高熔点和低疏水性(MP=275◦C,LogP=0.5),已被证明具有低水溶性。高熔点主要是由于亚胺官能团形成氢键能力较强,从而导致固体状态下强烈的分子间内聚力。通过用甲基取代亚胺的N-H供体基团,消除了亚胺-亚胺的氢键相互作用12,提高了热力学溶解度,并由于熔点降低而改善了物理化学性质。
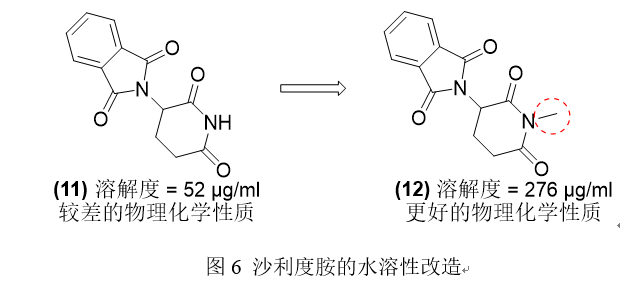
- 案例2:
化合物13被发现对鱼藤酮的毒性具有细胞保护活性,但水溶性差。周和他的同事们试图通过引入-CF3取代的苯环14来改善溶解度。进一步加入各种烷基后,溶解度没有改善,结论是加入-CF3取代的苯环后观察到的溶解度的改善可能是由于水和氟原子之间的氢键形成。化合物14表现出更好的热力溶解度,显著的细胞保护作用和更高的代谢稳定性。
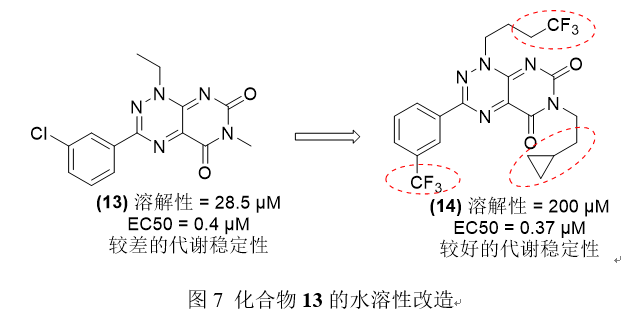
3.3通过前药修饰提高化合物水溶性
前药策略是克服药物发现中的各种障碍,包括溶解性差等问题,并且已经得到了广泛的文献报道。前体药物是一种非活性化合物,它以受控或可预测的方式在化学或酶作用下代谢成体内的母体活性形式。前药策略不改变原药结构,只靠添加助溶基团来提高水溶性,这是前药策略的一个优势。用于改善水溶性的前药设计有很多不同的方法,包括:磷酸化、氨基酸酯、糖基化、羧酸酯化、酰胺化和水溶性聚合物等。
- 案例1:
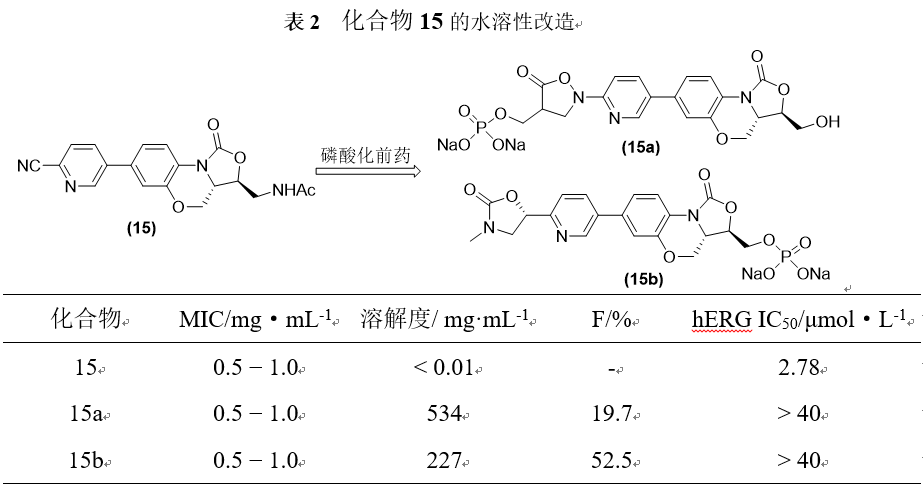
中国科学院上海药物研究所的杨玉社研究员课题组对具有抗耐药金葡菌(MRSA)的化合物15尝试了利用磷酸前药修饰来改善其水溶性。化合物15a和15b使用了两种不用的修饰方式,均具有与先导化合物相当的抗菌活性,水溶性提高了几万倍,同时对hERG通道的抑制活性IC50达到40µmol·L−1以上。化合物15b的生物利用度为52.5%。
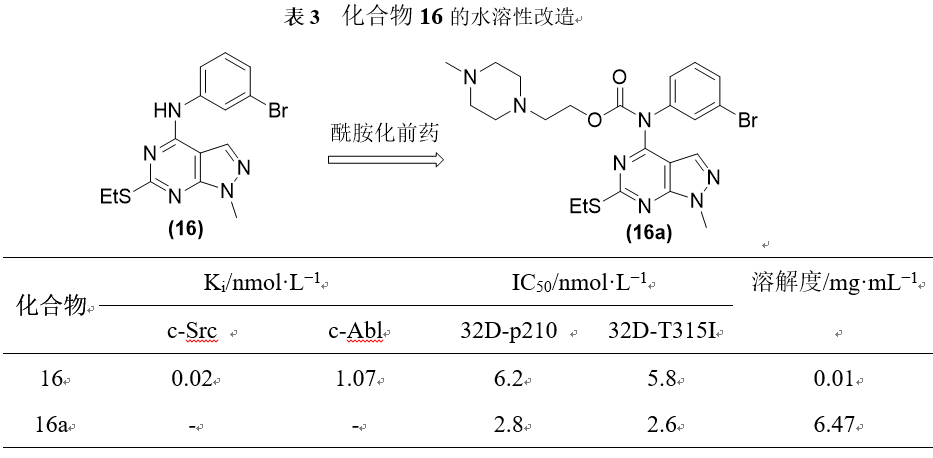
酰胺修饰的前药化合物16是一个Src/Abl双重抑制剂,在Src和Abl两种酶水平均具有纳摩尔的活性,但细胞活性并不高,究其原因很可能水溶性较差(仅为0.05mg·mL−1)。对16进行酰化的前药修饰后,化合物16a的溶解度提高了600倍,而相应的细胞水平活性也有了显著的改善。
- 案例2:
- 案例3:
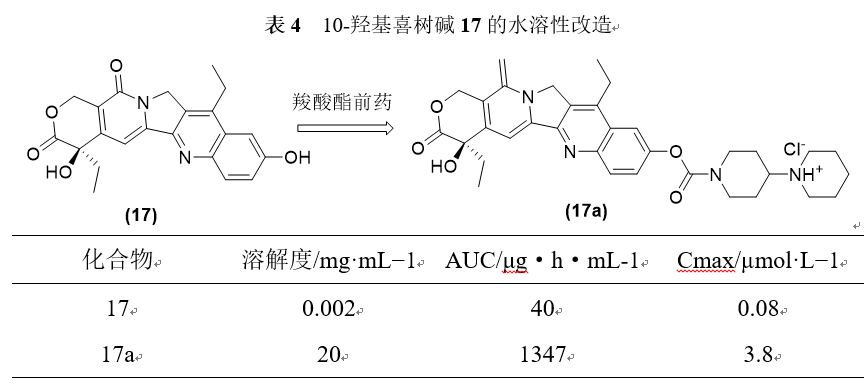
羧酸酯类前药是常用的前药设计方法之一。羧酸酯制备方便,体内水解容易,可以方便地引入水溶性基团。伊立替康17a的水溶性为20mg·mL−1,相比10-羟基喜树碱17本身(水溶性为2µg·mL−1)提高了上万倍。从药代力学参数看,药物的达峰浓度和暴露量分别提高了30~40倍以上。
3.4破坏分子的平面性、对称性和晶体堆积,以提高溶解度。
通过破坏分子的平面性和对称性来改善药物分子的水溶性是小分子药物发现过程中的一种新方法,这是一种替代传统方法的策略,如添加亲水基团或减少疏水性,这些传统方法通常会导致生物活性降低。已经观察到,紧密的晶体堆积也是先导化合物溶解性差的原因,紧密的晶体堆积和分子的平面排列可能会导致药物分子与水的相互作用较差,也会使化合物熔点升高,从而降低了溶解性。通过破坏晶体堆积或干扰分子平面性的化学修饰可以削弱稳定晶体化合物的分子间相互作用,从而提高其溶解性。
- 案例1:
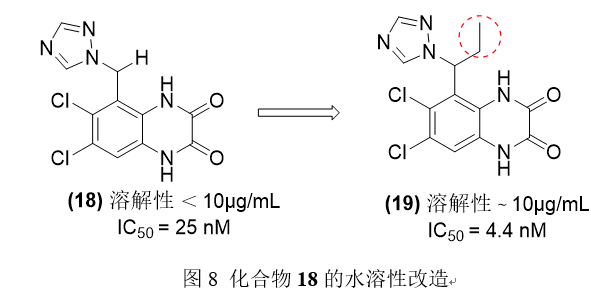
喹啉二酮衍生物18对NMDA受体有良好的活性,但水溶性差。为了改善水溶性,Fray和他的同事们对其进行化学修饰,在分子中插入一个乙基,使化合物19具有非平面构象,破坏了晶体堆积,并提高了热力学溶解度。
- 案例2:
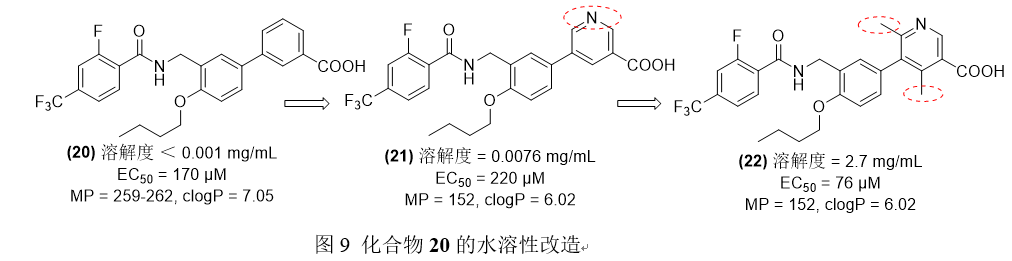
联苯羧酸衍生物20被发现对PPARγ具有部分激动/拮抗活性,PPARγ是一种配体激活转录因子,与脂肪酸代谢和细胞对胆固醇的吸收有关,但联苯羧酸衍生物的水溶性较差。Kasuga和他的同事们通过两种方法来改善热力学溶解度,一种是用生物等位吡啶核取代苯环,得到化合物21;另一种是通过添加取代基来扰乱平面性和晶体堆积,得到化合物22。[6]
3.5生物电子等位取代法改善溶解度。
生物电子等位取代是改善先导化合物溶解度、选择性和其他药学性质的经典和最常用的方法之一。生物电子等排体可分为两类,即经典生物电子等排体和非经典生物电子等排体。用吡啶、噻吩环或任何其他杂环生物等位取代先导化合物的苯环,可以改善溶解性和其他药物性质。
- 案例1:
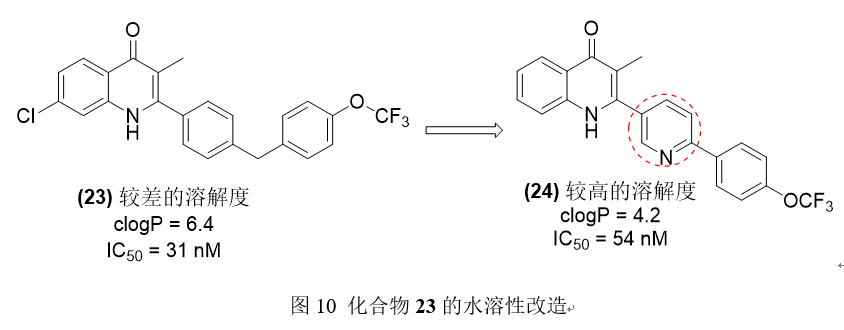
CK-2-68(23)是一种对恶性疟原虫II型具有良好亲和力的新分子,但由于较高的logP值和较差的水溶性,使得该分子受到药代动力学限制。CK-2-68核心为喹诺酮类,侧链为苯基。为了降低分子的logP和改善其水溶性,用不同的杂环体系对苯基侧链进行了多次生物电子等排替代,最终得到了一个新的具有吡啶侧链的分子SL-2-25(24)。吡啶喹诺酮类化合物有助于提高热力学溶解度,对恶性疟原虫具有良好的活性,并将logP从CK-2-98的6.4降低到SL-2-25的4.2。
- 案例2:
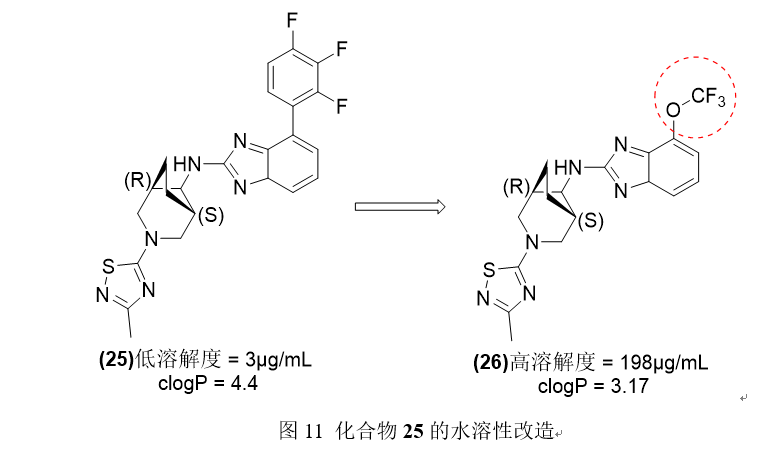
一种具有氟化苯基侧链的γ-分泌酶调节剂苯并三唑衍生物25的药代动力学不佳,水溶性和低IC50值。Sarmiento和他的同事试图通过结构上的修改来改善先导化合物的药代动力学和溶解性,其中之一是用-CF3烷氧基取代氟化苯环的非传统生物替代方法。与母体化合物相比,这种修饰导致了热力学水溶性增强,亲脂性降低,并提高了活性。
四、总结
水溶性在药物开发过程中的重要性无需多言,通过化学修饰手段改善水溶性的成效也显而易见。化学修饰的具体方法是多种多样的,但从理论策略层面均可归类于改善晶格能和水合能这两个方面,并可以从化合物的熔点、logP、构象等性质的变化趋势做出预测。充分理解和掌握这些策略并对症下药,将对指导先导化合物的优化起到事半功倍的效果。
参考文献:
[1]林瑞来. 难溶性药物溶解度的提高方法[J].中国医药指南, 2010 (3): 32-33.
[2]Das B, Baidya A T K, Mathew A T, et al. Structural modification aimed for improving solubility of lead compounds in early phase drug discovery[J]. Bioorganic & Medicinal Chemistry, 2022: 116614.
[3]Bhattachar S N, Deschenes L A, Wesley J A. Solubility: it's not just for physical chemists[J]. Drug discovery today, 2006, 11(21-22): 1012-1018.
[4]郭宗儒. 药物分子设计的策略: 分子的宏观性质与微观结构的统一[J]. 药学学报, 2008, 43(3): 227-233.
[5]栗增,王江,周宇,等. 先导化合物结构优化策略 (三)——通过化学修饰改善水溶性[J]. 药学学报, 2014, 49(9): 1238-1247.
[6]Kasuga J, Ishikawa M, Yonehara M, et al. Improvement of water-solubility of biarylcarboxylic acid peroxisome proliferator-activated receptor (PPAR) δ-selective partial agonists by disruption of molecular planarity/symmetry[J]. Bioorganic & medicinal chemistry, 2010, 18(20): 7164-7173.
<END>



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论