

































一、概述
1.仿制药是指与持有专利的原研药剂量(工艺装量)、安全性(已有大量临床数据支撑,符合人类使用)和效力(在同等剂型上使用)、质量(相同工艺下要达到原研药的用药质量标准或更好)、作用以及适应症(对临床药理要一样)上相同的一种仿制品。原研药专利到期以后,其他国家和制药厂即可生产仿制药。
2.仿制药并不是山寨药,在国际市场上,各国对仿制药都有着同样严格的要求;仿制药是与原研药具有相同的活性成分、剂型、给药途径和治疗作用的药品。进行质量和疗效的一致性评价,就是要求对已经批准上市的仿制药品,在质量和疗效上与原研药能够一致,在临床上与原研药可以相互替代,这样有利于节约社会的医药费用。
3.国家关于仿制药革新工作历程:
①2016年3月6日,国务院办公厅日前印发《关于开展仿制药质量和疗效一致性评价的意见》,要求化学药品新注册分类实施前批准上市的仿制药,凡未按照与原研药品质量和疗效一致原则审批的,均须开展一致性评价。
②国家基本药物目录(2012年版)中2007年10月1日前批准上市的化学药品仿制药口服固体制剂,应在2018年底前完成一致性评价。化学药品新注册分类实施前批准上市的其他仿制药,自首家品种通过一致性评价后,其他药品生产企业的相同品种原则上应在3年内完成一致性评价;逾期未完成的,不予再注册。
③2019年1月,国家卫健委、国家发改委等12部门联合发布《关于加快落实仿制药供应保障及使用政策工作方案》提出,2019年6月底前,发布第一批鼓励仿制的药品目录,引导企业研发、注册和生产。根据临床用药需求,2020年起,每年年底前发布鼓励仿制的药品目录。
4.仿制药也有创新点,比如工艺创新(设备/设施改进创新、实用型创新)、质量分析方法创新、成本控制等。
5.仿制药(制剂)在研发阶段工作可以分为:项目调研、处方研究、小试处方工艺筛选、放大及中试研究、可行批生产研究、工艺验证、注册资料整理、质量标准复核、CDE沟通、注册核查等工作。
6.本文主要从上述各部分内容进行详细剖析,着重讲解关键点、重点。
二、研发主要工作内容剖析
§项目调研
1.调研的内容要全面、精细、时间长,形成调研报告;同时形成项目可行性报告。
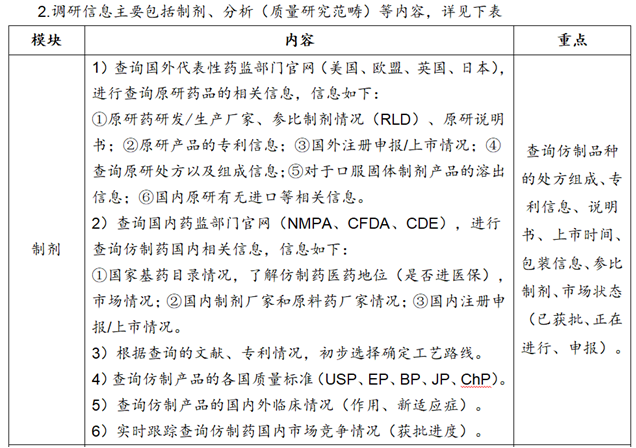
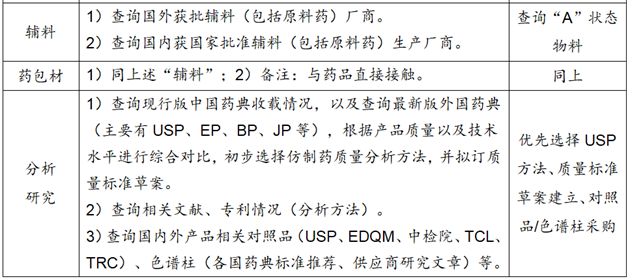
3.附件信息列表
①质量标准草案:原料药、辅料、药包材、制剂等。
②分析方法:有关物质、含量、其他杂质(残留溶剂、基因毒杂质、无机盐杂质)等。
③对照品:杂质对照品、主成分对照品、研究杂质等。
④色谱柱:有关物质(杂质分离)、含量、残留溶剂等LC/GC/IC色谱柱。
⑤处方查询:FDA的Reviews-https://dailymed.nlm.nih.gov/、欧盟EMA的Product information、日本PMDA的IF文件、英国EMC的SPC、中国的NMPA/处方集、阿根廷/巴西/西班牙药监局等。
⑥参比制剂查询:美国FDA-https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm。中国NMPA-https://www.nmpa.gov.cn/datasearch/search-result.html。
网站-FDA橙皮书/参比购网-https://www.canbigou.com/directory.html。
⑦说明书查询:https://dailymed.nlm.nih.gov/dailymed/、药融云数据库等。
⑧药品标准查询:https://www.drugfuture.com/standard/。
§处方研究
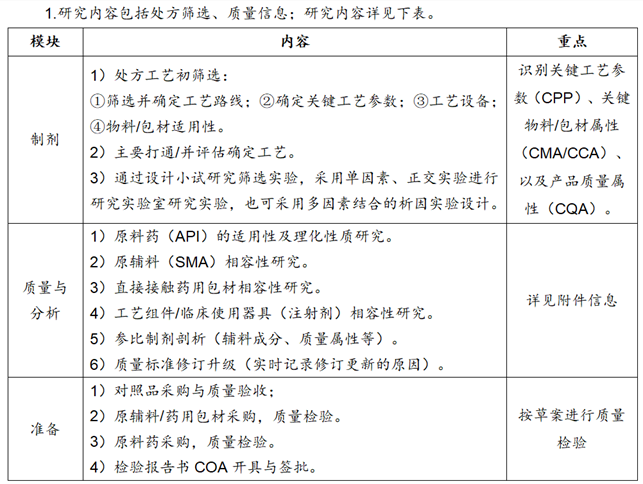
2.附件信息
①原料药适用性及理化性质研究:主要通过选择2-3家通过注册审批为A状态的厂家的上市生产的原料药小样,进行自检对比质量,以及相关稳定性研究数据对比评估;选择制剂用原料药(评估确定1-2个合格供应商)。
②包材/工艺组件相容性研究:质量研究内容,可在稳定性研究一起进行;主要关注点抗氧剂、硫化促进剂、脂肪酸、塑化剂、烷烃及无机杂质、重金属等。
③原辅料相容性研究:主要考察原辅料/辅料对主要的影响(是否发生化学/生物反应,导致主要有关物质杂质个数/杂质含量增加);考察是否影响成品制剂有关物质(杂质分离效果-辅料多少)/含量检测。
④参比制剂剖析:口服固体制剂(片剂/胶囊)-溶出度/主药含量研究;注射剂-装量研究等;以及稳定性考察研究。参比制剂要全检(选择2-3批次新生产批号)。
备注1:参考指导原则《普通口服固体制剂溶出度试验指导原则》、《普通口服固体制剂溶出曲线测定与比较指导原则》、JP《仿制药生物等效性试验指导原则2012版》、USP《口服缓释制剂体内外相关性研究技术指导原则》等等。
备注2:参比制剂剖析质量检验,一般参照进口标准/原研标准或现行中国药典标准,前后研究所用方法应一致。
§小试处方工艺筛选(小试研究)

2.附件信息
①处方工艺筛选:按照制剂处方常规用量(辅料)以及常规工艺/工艺路线/工艺参数,进行小试实验,参照制剂基本性能(中间产品颗粒可压性-片剂,流动性-片剂/注射剂/胶囊剂,硬度/脆碎度-片剂,水分)等指标进行筛选实验,并对比质量;例1片剂需要进行大量溶出曲线实验,选择突变(有区分力)溶出曲线与原研制剂/参比制剂进行比较,从而找出与原研制剂的区别,通过调节辅料用量或选择新型替代辅料,使溶出曲线达到参比一致。例2注射剂需要进行装量研究,提供调节生产设备控制填装量,使临床使用疗效达到一致。
②预影响因素/预加速对比试验:通过筛选确认2-3个工艺产品进行影响因素(高温、高湿、强光)/预加速(40℃-75%,1、2、3月)对比试验;选出合适工艺。
③连续3批次小试:通过上述①/②筛选工艺连续3批工艺实验,最终产品收率是否预期、质量是否合格平行等。
④质量标准与工艺:1)建立中间产品质量标准(成品前一步产品质量)、制剂成品质量,主要是为中试/可行批质量检测准备,实时记录修订升级原因;2)筛选并确认2-3个评估后的最佳处方工艺,分别进行实验室小试生产出小样,和原研制剂/参比制剂进行质量对比(影响因素研究、辅料/原辅包材相容性研究试验),选择主药敏感的因素进行。
§放大及中试研究

2.附件信息
①中试放大研究:对照工艺/工艺路线/工艺参数进行考察研究工艺稳定性,以及生产设备的适配性(包括生产设备调试、技改),进行3批次放大中试生产,制订生产工艺规程、批生产记录;实时记录收率、物料平衡、损失率,为工艺验证作指标。
②连续3批次中试放大质量:质量检测,质量评估并开具签批合格COA。
③分析方法验证:产品检测分析方法(有关物质、含量、限度检查、无菌/微生物限度/细菌内毒素)、设备清洁方法等验证。
§可行批生产研究
可行批生产研究主要考察包括生产设备调试、技改后的工艺稳定性,以及生产设备的适配性;进行1-2批次;质量检验合格,同时形成可行批报告。
§工艺验证
1.工艺验证前准备:1)物料准备(包括包材/包装);2)检测用物料准备(色谱柱、对照品/标准物质、检测试剂等);3)产品质量标准/检测分析方法是否准备验收完毕;4)生产批记录/检测批记录准备/清洁验证记录等;5)工艺风险评估等。
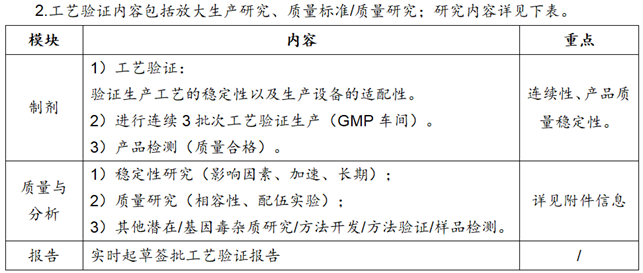
3.附件信息
①工艺验证:按照批准生效的工艺规程进行工艺验证生产3批,验证包括生产设备调试、技改后的工艺与质量稳定性,是否达到验证方案目标。
②质量检测:质量评估报告并开具签批合格COA。
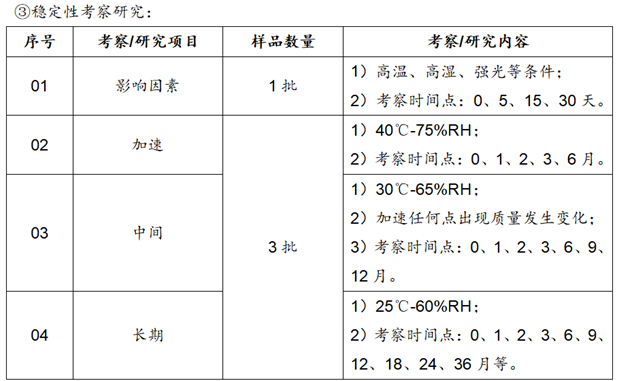
④其他潜在/基因毒杂质研究:工艺验证3批均未检出或检出量都低于限度的1/3,可不纳入产品质量标准中;但是在加速和长期稳定性考察末期,应进行检查。
⑤相容性研究:包材/工艺组件相容性研究实验;与参比制剂一起进行对比实验。
⑥配伍研究:主要考察说明书以及临床使用说明书进行模拟配伍质量实验。
§注册资料整理
1.主要资料整理:1)稳定性考察数据汇总整理与总结;2)工艺控制、质量控制资料;3)产品质量标准研究与质量研究资料等。
2.内容有:药学研究资料综述、证明性文件资料、生产工艺研究资料及文献、处方及工艺研究资料与文献、质量研究资料、生产信息、质量控制(辅料与成品质量控制)、药效学(药理毒理)研究资料、对照品与杂质谱研究资料、稳定性研究资料等。
§质量标准复核
1.提交药品注册检验申请表/注册申报受理通知书等。
2.样品及物品准备:工艺验证3批样品(3倍检验量,至少还有6个月有效期)、对照品(杂质/主成分对照品,3倍检验量)、相关色谱柱等。
3.文件资料:1)相关原料药、原辅包材证明性文件资料;2)质量标准(最新版)及质量标准对比表;3)相关检验方法分析方法验证报告;4)3批工艺验证样品企业自检COA。
4.全部纸质资料文件均需盖企业章(准备电子版1份)。
§注册现场核查
1.注册核查:1)注册研发现场-药学研制/药理毒理学研制/药物临床试验;2)生产现场。
2.目的:核实注册申报资料的真实性、一致性以及药品上市商业化生产条件。
3.重点:申报资料、工艺验证、质量控制、生产现场、辅助文件、人员培训等。
三、总结
1.仿制药是和被仿制产品含有相同的活性成分及辅料(包括直接接触的包材);和被仿制产品的临床适应症、剂型、规格、给药途径一致,生物等效,且产品质量同等一致的最基本要求;对于仿制药,不需要重复进行创新药批准之前进行的多年临床前动物研究和人体临床研究,而是通过证明和原创药的生物等效性即可获得批准;仿制药是与被仿制药具有相同的活性成分、剂型、给药途径和治疗作用的替代药品,具有降低医疗支出、提高药品可及性、提升医疗服务水平等重要经济和社会效益。
2.对于口服制剂,除了上述共性外,仿制药一致性评价的重点工作是使仿制药与原研/参比制剂在体内外的溶出曲线,速度达到一致,需要做BE试验。
3.对于注射制剂,除了上述共性外,注射剂药物由于其特殊性(直接注入人体血液中),所以注射剂评价的重要内容是对于其安全性研究;杂质,稳定性,无菌保障,包材相容性等研究是必须要进行的,还有特别要求进行临床研究等。
4.仿制药注册申报全流程环环相接,一步一步向前推进,着重注意产品质量和申报单位生产条件是否具备上市生产硬性要求。
<END>



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论