


































在现代人类医学的宏大图景中,仿制药构成了全球医疗卫生体系运转的最重要基石。仿制药以其亲民的价格和极高的可及性,承担了绝大部分基础医疗与慢病管理的重任,极大地缓解了各国的公共卫生财政压力。然而,仿制药替代昂贵原研药的核心前提,在于一个看似极其简单、实则在科学界充满争议与挑战的命题:如何从科学意义上证明“两者完全相同”?
生物系统本身是一个极其精密且充满非线性和个体间巨大差异的黑箱。因此,在复杂的生物学和生理学意义上,定义并证明两种化学有效成分相同,但其生产工艺、辅料配方、甚至晶型结构可能迥异的药物制剂,能够在人体内展现出丝毫不差的临床疗效与安全性,是一项极度艰巨的系统工程。
生物等效性(Bioequivalence, BE)研究是现代药物评价体系中的核心组成部分,主要用于证实仿制药与原研药在人体内的吸收速率与吸收程度的一致性。本文基于药代动力学(Pharmacokinetics, PK)与统计学原理,系统梳理了BE概念的历史演变,深入探讨了不同理化特性及药代动力学特征药物(如高变异药物、窄治疗指数药物等)的特殊评价策略,并对BE试验的统计学判定标准及质量管理体系(GCP/GLP)进行了全面解析。
一、药物等效性评价的科学起源与核心概念
在现代药代动力学学科建立初期,临床医学界普遍存在一种认知偏差,即认为含有相同化学成分的药物必然具有相同的临床疗效。然而,20世纪60至70年代的临床研究文献指出,不同制药企业生产的同种化学药物(如氯霉素、保泰松、四环素和土霉素等)在实际应用中,其血药浓度表现出极大的差异。这种药代动力学特征的波动,不仅可能导致临床治疗失败,更可能增加患者发生毒性反应的风险。
20世纪70年代爆发的“地高辛(Digoxin)危机”,直接逼迫美国食品药品监督管理局(FDA)从根本上重塑了现代药品的监管体系。地高辛是从洋地黄植物中提取的一种强心苷类药物,广泛用于治疗充血性心力衰竭和心房颤动,是当时心血管疾病管理中不可或缺的救命药。然而,地高辛具有一个致命的特点——它的治疗指数极窄。该药的最小有效浓度与中毒浓度之间的界限微乎其微(治疗浓度通常在1-2 ng/mL),极微小的剂量波动不仅不能救命,反而会迅速引发心律失常、严重心动过缓、心脏传导阻滞甚至死亡。
地高辛的作用机制
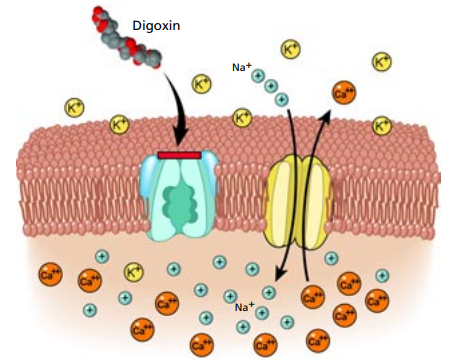
1970年,美国食品药品监督管理局(FDA)针对不同企业生产的地高辛片剂进行了一项系统的血药浓度检测研究,结果表明,不同厂家甚至同一厂家不同批次的制剂,其峰浓度差异最高可达7倍。科学界由此认识到,药物制剂中的辅料成分、粉体学性质(如粒径大小)、崩解时间及溶出度等制剂工艺参数,均可能深刻影响药物在体内的暴露水平。基于这一背景,药物等效性评价体系应运而生,其核心概念包含以下三个递进的维度:
① 药学等效性(Pharmaceutical Equivalence):指不同药物制剂含有相同摩尔剂量的同种活性成分,具有相同的剂型和给药途径,并且在规格、质量、纯度及成分鉴别等方面符合相同的药典或其他适用标准。然而,药学等效并不等同于体内行为一致,辅料与工艺的差异仍可能改变药物的释放与吸收动力学。
② 生物等效性(Bioequivalence, BE):指将两种药学等效的制剂以相同摩尔剂量给药后,其活性成分到达药物作用靶部位的吸收速率和吸收程度不存在显著的统计学差异。
③ 治疗等效性(Therapeutic Equivalence):当两种药学等效的药物被科学证实具有生物等效性时,即可被认定为具有治疗等效性。在按说明书规定途径给药的前提下,治疗等效的药物应表现出完全相同的安全性与有效性,进而在临床实践中具备可替代性(互换性)。
二、生物等效性评价体系的历史沿革
生物等效性的评价方法并非静态不变,而是随着生物药剂学、统计学与分析化学的发展经历了三个重要阶段的演进:
(一)第一阶段:20世纪70至80年代(概念确立与基础构建)
在BE建立的初期,由于早期的上市标准无法有效筛选出存在生物不等效风险的仿制药,导致部分治疗失败或毒副反应案例的发生。这一阶段的主要任务是明确临床重要性高、治疗浓度与毒性浓度接近的特定药物类别,并探索能够有效预测人体生物利用度(Bioavailability, BA)的体外溶出方法与动物模型。
(二)第二阶段:20世纪90年代(理论深化与模型引入)
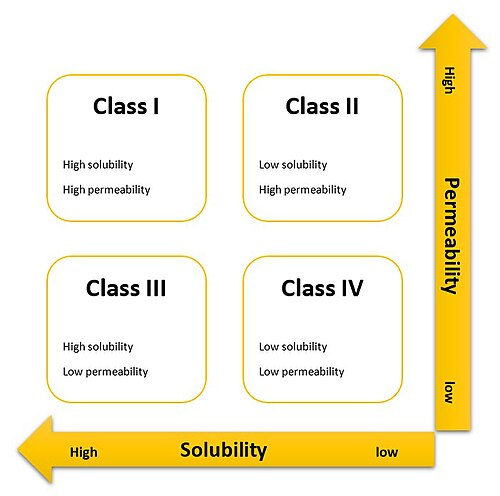
这一时期,BE的评价理论得到显著丰富。科学界提出了个体生物等效性(Individual Bioequivalence, IBE)的概念,旨在比较同一受试者对不同制剂的个体内变异,以此为仿制药在个体层面的可互换性提供更充分的理论依据。同时,生物药剂学分类系统(Biopharmaceutics Classification System, BCS)及房室吸收与转运(CAT)模型被正式提出并应用于BE指导原则中。尽管IBE在理论上更为严谨,但受限于试验设计成本及统计学方法的复杂性,平均生物等效性(Average Bioequivalence, ABE)目前仍是评价的主要标准。
BCS分类
(三)第三阶段:21世纪伊始至今(精准评价与复杂制剂策略)
进入21世纪,随着仿制药种类的多样化,监管机构针对特殊药代动力学特征的药物(如高变异药物、长半衰期药物、窄治疗指数药物及局部用药)建立了一系列更具针对性的BE评价体系与豁免标准。
三、基于药物特性的差异化 BE 评价策略
现代药物评价要求结合药物的理化性质与体内处置特征,制定科学、合理的等效性试验方案。基于药物的溶解性、渗透性及主要清除途径,不同类型的药物采取了不同的评价策略。
(一)BCS I类药物的豁免机制
对于高溶解性、高渗透性(BCS I类)的速释口服固体制剂,若其在胃肠道环境中稳定、处方中的辅料不显著影响吸收、非窄治疗指数药物,且能在规定的体外实验条件下快速溶解,则通常认为其体外快速溶出足以保证体内快速吸收。对于此类药物,监管机构允许其申请豁免体内BE试验。
(二)高变异药物(Highly Variable Drugs)的统计学放宽
高变异药物指受试者药代动力学参数(主要为吸收速率指标Cmax)的个体内变异系数(CV)≥30%的药物。在常规的等效性判定标准下,高变异药物即便与自身参比,也具有极高的等效性检验失败率。为科学评价此类药物,通常采用三交叉或四交叉的重复试验设计,以准确评估其个体内差异。对于安全性良好、治疗窗较宽的药物,基于公众用药安全的前提,可采用参比药品标度的平均生物等效性(Reference-Scaled Average Bioequivalence, RSABE)方法。该方法允许根据参比药品的个体内变异值,将传统的80.00%~125.00%等效区间进行科学拓宽,Cmax的接受限度最宽可放宽至69.84%~143.19%。这在保证有效性的同时,显著降低了不必要的人体临床试验暴露。
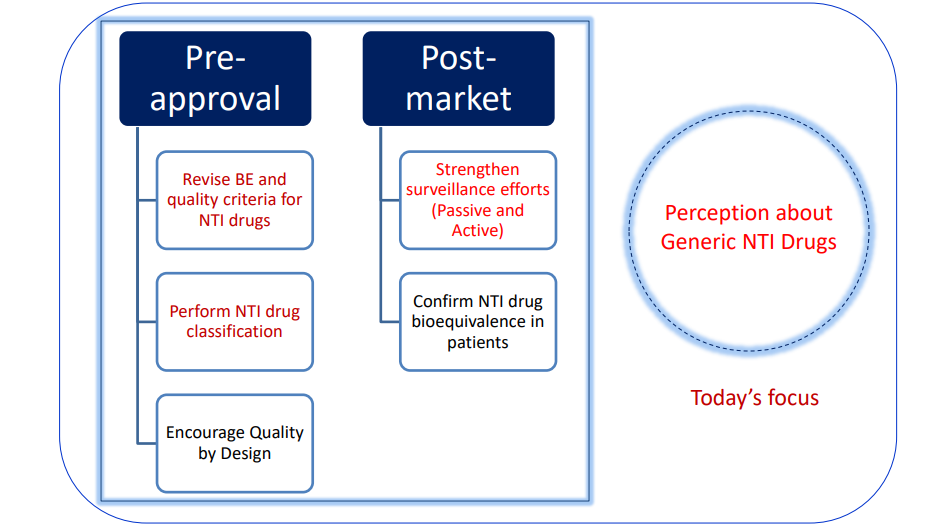
(三)窄治疗指数(Narrow Therapeutic Index, NTI)药物的严格界定
与高变异药物相反,窄治疗指数(NTI)药物的治疗剂量与中毒剂量极度接近,血药浓度的微小波动(如临床实践中低于20%的剂量微调)即可引发严重的治疗失败或毒副反应。此类药物通常需要进行治疗药物浓度监测(TDM)。针对NTI药物,等效性评价的宽容度被大幅收紧。其血药浓度-时间曲线下面积(AUC)的可接受置信区间必须缩窄至90.00%~111.11%。在峰浓度(Cmax)对药物安全性或有效性起决定性作用的情况下,Cmax同样适用90.00%~111.11%的严格限定。
FDA努力确保仿制NTI药物的安全性和有效性
(四)长半衰期药物的局部吸收评价
对于半衰期极长的常规剂型,法规通常规定采样周期不需超过72小时即可完成等效性评价。然而,对于呈现多相药代动力学行为的缓释或控释制剂,总体的AUC和Cmax可能掩盖药物在特定时间段内吸收特性的差异。因此,评估此类制剂的等效性时,常需引入部分药—时曲线下面积(partial AUC, pAUC)作为附加的药代动力学参数,以确保受试制剂在临床相关的特定时间间隔内,其到达作用部位的速率与程度同参比制剂保持一致。
(五)局部作用药物的终点替代
局部作用药物(如皮肤外用药、胃肠道黏膜保护剂、吸入剂等)通过在作用部位直接起效发挥作用,并不依赖于全身血液循环的药物浓度。事实上,局部用药的系统吸收率增加,可能反而预示着靶部位药物浓度的降低与疗效的衰减。由于血药浓度无法客观反映此类药物的疗效,传统的药代动力学评价方法存在局限性。对此,监管机构通常要求采用临床疗效终点作为等效性评价指标。例如,抗溃疡药硫糖铝需通过针对十二指肠溃疡患者的8周临床疗效试验来判定等效性 ;对于局部皮肤外用药,目前学术界亦在探索体外扩散法、微透析技术及近红外光谱技术等新方法用于局部药物浓度的精确测定。
四、 生物等效性判定的统计学原理与模型
BE评价本质上是基于药代动力学参数的统计学推断。其核心目的是利用样本的统计特征,以特定的置信度推断受试药品(T)与参比药品(R)的总体参数分布差异是否在预设的临床等效可接受范围之内。
在进行统计分析时,首先需对主要的药代动力学参数(AUC、Cmax等)进行对数转换,随后采用多因素方差分析(Analysis of Variance, ANOVA)进行显著性检验。ANOVA的目的是分解并量化试验数据中的误差来源(包括制剂间、个体间、周期间以及服药顺序间的差异),为后续的检验提供均方误差(Mean Squared Error, MSE)值。
生物等效性判定目前国际公认的唯一标准为双单侧t检验(Two One-Sided Tests, TOST)及(1-2α)%置信区间法。与传统的差异性检验不同,等效性检验设定的无效假设为“两药不等效”(即受试药品的参数均值超出了参比药品的高限和低限)。通过在上限和下限两个方向分别进行单侧t检验:
① 验证受试药品均值在统计学上没有显著高于参比药品均值的125.00%(P<0.05)。
② 验证受试药品均值在统计学上没有显著低于参比药品均值的80.00%(P<0.05)。
当且仅当这两个单侧检验均被证实,即经对数转换后的受试药品参数均值的90%置信区间(90% CI),完全落在参比药品均值的80.00%~125.00%范围内时,方可以90%的概率拒绝无效假设,作出两制剂生物等效的科学推断。
五、 临床试验与生物分析的质量管理体系
生物等效性研究的科学性与合法性,建立在极其严密的数据真实性与过程合规性之上。任何一项BE试验的开展,必须严格遵循《世界医学大会赫尔辛基宣言》的伦理原则,并在法规层面全面契合《药物临床试验质量管理规范》(GCP)以及《药物非临床试验质量管理规范》(GLP)的要求。此外,世界卫生组织(WHO)发布的《药物临床/非临床试验质量管理规范》(GCLP)进一步强化了对临床生物样本检测数据的监督,确保实验室数据的可靠性与完整性。
为了实现这一目标,临床研究机构及分析实验室必须建立系统、详尽的标准操作规程(Standard Operating Procedures, SOP)体系。

SOP涵盖了从试验准备到报告出具的全部细节,构成了质量控制与质量保证的基石。其关键环节包括但不限于:
① 伦理与受试者保护:包含知情同意书的设计与签署程序(SOP 8, 12)、受试者的招募、入组筛查(SOP 29, 30)以及不良事件(AE)与严重不良事件(SAE)的应急处理与规范报告(SOP 18, 40)。
② 试验用药品的严密闭环:涉及试验用药品(Investigational Product)的接收、储存温湿度监控、精确发放、定点回收及最终的规范处置(SOP 22-27),以杜绝交叉污染与使用差错。
③ 生物样本的流转规范:规定了从临床血样/尿样的定时采集、离心处理、低温保存,到样本编码与跨区域冷链转运的标准流程(SOP 44-49),以确保生物分子的稳定性。
④ 生物分析方法的确证:分析实验室必须出具严密的分析方法验证数据,包括标准曲线的建立、定量下限(LLOQ)的确认、选择性、准确度、精密度、基质效应考察及稳定性测试(SOP 53),并确立图谱积分规则及分析批次接受的统计学标准(SOP 54, 55)。
所有试验过程中产生的数据均须作为原始数据予以妥善保存,并接受申办方、独立稽查部门乃至国家药品监督管理部门的严格核查。
六、结语
综上所述,生物等效性(BE)研究是一项融合了临床医学、药代动力学、生物分析化学及数理统计学的综合性系统工程。它通过严密的法规约束与科学的统计推断,摒弃了早年仅凭“化学成分相同”即认定“疗效一致”的粗放式管理。随着对药物吸收机制认知的深化,BE评价标准正逐步向基于分子靶点分布及局部药代动力学的精细化方向演进。这套高度标准化的评价体系,不仅为仿制药的有效性与安全性筑起了坚实的科学壁垒,更为全球范围内基本药物的可及性与可互换性提供了无可替代的技术保障。
扩展阅读:
1. 微观视角下的药物研发:原料药固态性质全解析与药企战略抉择
2. 解锁口服药物吸收密码:溶解性、渗透性及S-P矛盾全解析
3. 抗抑郁药沃替西汀研发全解:多模式机制、晶型奥秘与早期原料药开发大揭秘
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论