
































杂质研究是药品研发的一项重要内容,它包括了选择合适的分析方法,准确的分辨、与测定杂质的含量并综合药物、毒理及临床研究结果确定杂质的合理限度。杂质研究贯穿于药品研发的整个过程。
制剂中的杂质来源主要有三种:第一种来源于原料药中的工艺杂质,该类杂质一般通过原料药进行研究与控制,当原料药中的杂质同时也是降解杂质时,需要在制剂中关注;第二种是制剂中活性成分由于自身结构原因降解得到的杂质;第三种是制剂中的活性成分与辅料之间相互作用后产生的杂质,其中还包括辅料中的杂质与药物相互作用后的降解产物。
1. 关于制剂活性成分降解
制剂中药物活性成分由于结构的特点,在某些环境条件下会发生降解,常见的降解方式有下面几种。
水解作用。当药物中含有酯、胺、内酯、内酰胺等结构,在储存条件或者湿度较高条件下易发生水解降解。
氧化作用。氧化是降水解反应之后的第二大降解反应。氧化降解的反应机制比较复杂,包括去除带正电原子、自由基、电子,或者加成带负电基团。氧化反应可以在有氧、重金属、光照条件下发生。
同分异构化作用。异构化包括光学异构化和位置异构化。对于某些药物来说,异构体在药理或者毒理中的活性与活性成分相比有很大的差异。
光降解作用。在光照条件下,一些氧化还原、基团中环转变和聚合反应时常会反应。
聚合降解。分子间的聚合也是活性成分常常发生的一种降解反应。
表1:常见药物活性成分降解方式
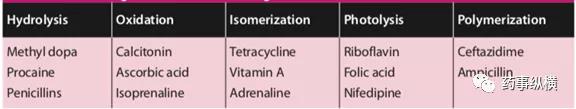
还有一些反应包括脱水作用、消除作用和成环作用。影响上述反应的主要因素为:温度、酸碱度(pH值)、水分、环境相对湿度、催化剂的存在、氧气、光照、物理形态及药物和辅料的粒度等。
2. 关于制剂活性成分与辅料的相容性
制剂降解杂质的主要原因是制剂中活性成分与辅料间的相互作用。大部分降解杂质是通过活性成分的水解、氧化或者活性成分与辅料中杂质的反应产生。
短期的相容性研究可以用来支持新药前期或者I期临床研究,对于II或者III期的临床研究,需要更加深入的辅料相容性研究,以保证药物制剂的安全性和稳定性。
辅料相容性研究之前,需要掌握了解原料药的理化性质和稳定性,包括原料药的合成工艺、原料药的在不同pH值、高温、光照条件下的溶液稳定性情况。
不同原料药的性质不同,在与辅料的相容性研究中需要单独设计。
辅料相容性研究的核心包括:混合样品设计和制备、混合样品的成分比例、储存条件和有稳定性指示的分析方法。在某些条件下可以加入一些活性杂质或者水分来加速与原料药反应,也可以使用不同盐型、晶型或无定型原料药进行相容性研究。
储存条件一般有常温、加速和光照降解条件。测试结果需要分析相容性样品的物理与化学的稳定性。辅料相容性研究用来支持制剂处方研究,以及制剂货架期预测。
表1:常见不相容性药物与辅料
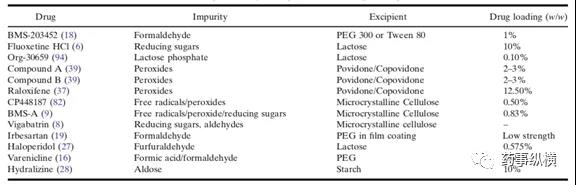
3. 关于制剂活性成分与辅料中活性杂质的相容性
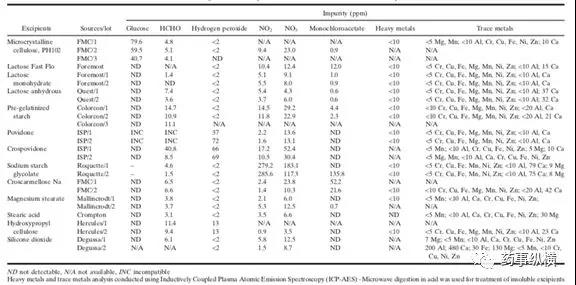
辅料自身含有的特殊杂质在药物相容性研究中需要密切关注。辅料中常见杂质有:乙醛、还原糖、过氧化物、硝酸盐、亚硝酸盐、金属及溶剂等残留。
这些杂质的来源是辅料生产或者储存过程中产生的,根据辅料中的杂质与原料药之间的相互作用,可以设计不同条件下的相容性实验,来判断原料药与辅料之间的相容性问题。
表2:辅料中的常见活性杂质
- 还原糖。葡萄糖和乳糖是还原糖的赋形剂。胺类药物与这些糖的不相容性是众所周知的。微量还原糖也存在于非还原性辅料中,如微晶纤维素、淀粉、甘露醇、麦芽糖醇和蔗糖。
还原糖与伯胺和肿胺类药物常见的反应为美拉德反应。糖基胺通常是还原糖中的糖基羟基被胺基取代反应的第一步。糖基胺经历重排产生酮胺,并与其他胺类进一步反应,产生高级美拉德反应产物,如黑色素,导致药物的变色。低载药量、高含水量和碱性微环境有助于加快美拉德反应速率。
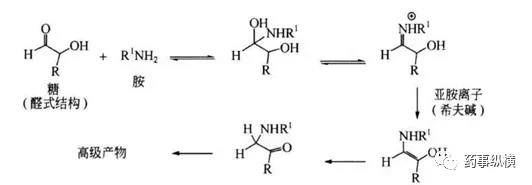
- 醛类杂质。甲醛、乙醛和糠醛(可能还有其他醛)是常见的醛杂质,它们存在于淀粉、预糊化淀粉、交联聚维酮、羟丙基纤维素、聚乙二醇和乳糖中醛类与活性成分中的伯胺、肿胺、酮基邻位碳易发生反应。如下图所示:、
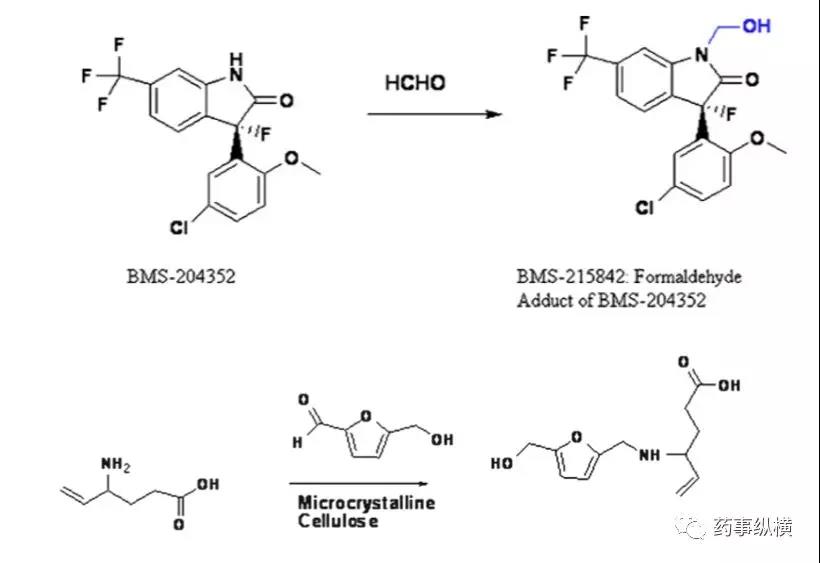
- 过氧化氢和过氧化物。药物中过氧化物的来源常见辅料如聚维酮、羟丙基纤维素、聚维酮、聚乙二醇、聚氧化乙烯和聚山梨酯。
通常过氧化物可以是有机过氧化物(ROOR′)或氢过氧化物(ROOH)。过氧化物可形成过氧自由基,然后过氧自由基通过另一反应物中提取氢自由基来形成过氧化氢,同时生成另一个碳自由基,从而参与自催化循环。
任何易氧化的药物都易与含过氧化氢的辅料(如PVP、交联聚维酮和HPC)发生相互作用。原料药表现出电子反应,如N-氧化物的形成和硫醇的氧化,或一个电子氧化,如苄基氢原子的失去。
- 硝酸盐和亚硝酸盐。硝酸盐和亚硝酸盐是常见的杂质,在大多数辅料中都能找到,含量为百万分之几。淀粉乙醇酸钠、交联羧甲基纤维素钠、预糊化淀粉、PVP、cPVP和乳糖是含微量硝酸盐或亚硝酸盐。
含氮药物化合物由于与辅料中的亚硝酸盐或硝酸盐杂质相互作用,有可能在药物产品中形成N-亚硝基化合物。能形成N-亚硝基化合物的官能团包括二烷基、烷基芳基、二芳基、环仲胺、N-烷基脲、N-烷基氨基甲酸酯和N-烷基酰胺,以形成亚硝胺或亚硝胺。氰胺、胍、酰胺、羟胺、肼、腙也可以形成N-亚硝基化合物。
- 有机酸:辅料中的一些有机酸如甲酸、乙酸、一氯乙酸常出现在辅料聚乙二醇,羟丙基甲基纤维素,聚维酮,聚乙烯醇,聚乙烯醇醋酸纤维,丁酸淀粉乙醇钠,交联羧甲基纤维素钠当中,如表3。
表3:含有机酸的常见辅料
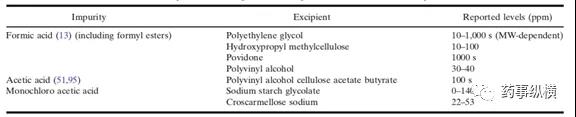
有机酸能够与药物中含胺基、羟基反应。也能与醇基反应成酯。研究者发现甲酸可以与药物中的肿胺发生N-甲酰化和N-甲基化反应。
4. 关于法规
ICH Q3B是新药制剂中杂质研究的基本指南。通常情况下,存在于原料药中的非降解杂质、辅料中的杂质或包装容器渗出的杂质,在制剂中不进行监控。新制剂关注生产过程及稳定性考察中所发现的降解杂质,有三限(报告限、鉴定限、界定限)要求,它的限度接受要求可参考下表。
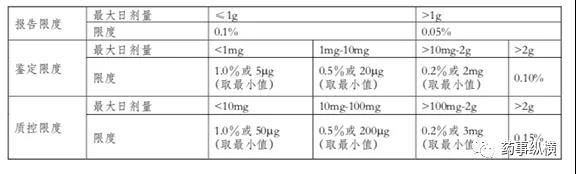
当制剂中降解产物的水平超过质控限度时,需要有杂质的安全性评价数据来支持其限度水平。如果降解产物本身是动物或人体中的重要降解产物,也可认为其已通过质控;根据科学原理和药品的类别及临床使用情况,某些新制剂降解产物的质控限度可以制定得更高或更低。原料药中的基因毒性杂质在制剂中也需要引起重视与关注。
制剂注册申报资料中涉及杂质研究的有:3.2.P.2.3生产工艺的开发中批分析,3.2.P.3.4关键中间步骤和中间体的控制,3.2.P.5.1制剂质量标准中杂质的限度,3.2.P.5.5制剂的杂质分析,3.2.P.5.4批检验报告,3.2.P.5.6质量标准制定依据,3.2.P.6对照品杂质对照物料。
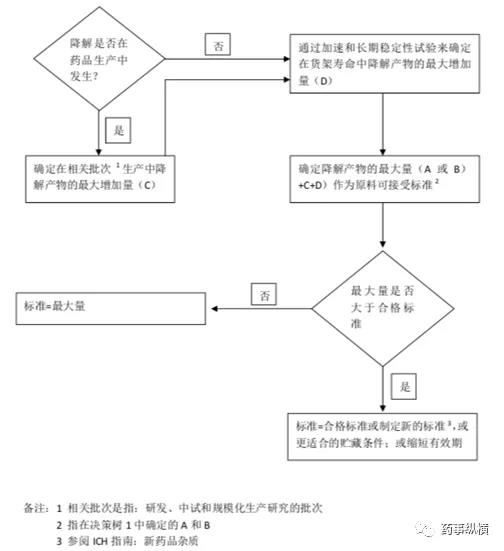
制剂质量标准制定时应包括在上市产品生产和推荐的贮藏条件下预期会出现的降解产物,这些降解产物可参考上市或拟上市生产工艺中发现的降解产生来确定。质量标准制定的决策树参考下图。
制剂中杂质标准制定决策树
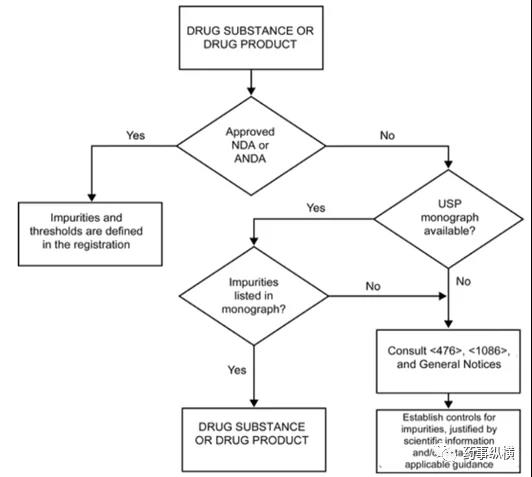
USP通则<1086, 原料药和制剂杂质>是原料药和制剂中杂质的研究与控制指南。制剂中杂质是制剂中降解的有机杂质。USP在2017年新建立了一个通则<476, 原料药与制剂的杂质控制>,主要内容是药典各论中已有的原料药与制剂中杂质分析测试与控制,提出了有机杂质控制决策树,如下图,具体内容可参考PF43(6)。
原料药和制剂中有机杂质控制决策树
5. 关于制剂常用分析方法
- 热分析(Thermalanalysis)
热分析是在升温过程中把辅料和有效成分的物理化学性质变化作为温度函数进行统计研究的方法,包括热重法,热差分析方法。
- HPLC法
HPLC方法是常用的药物制剂杂质研究分析方法。通过HPLC方法可以测定制剂中主药与辅料混合物有效成分的含量变化来确认药物是否降解。
- XRPD法
X射线粉末衍射法常用于监测制剂中药物与辅料的形态变化、晶形转化以及水合变化等程度。它与热分析法相结合使用,能够有效判断药物和辅料间的相容性。
- FTIR法
傅里叶变换红外色谱法(FTIR)可以得到原料药,、主药与辅料混合物的红外光谱图,然后通过光谱反映的信息得出是否出现了新物质。FTIR可用于药物与辅料的相容性研究。
6. 关于其它特殊药物制剂杂质
- 非处方药物:与FDA的新药申请和ANDAs不同的是,OTC药品获准上市之前,不会受到FDA的单独质量评估。为了解决对OTC产品质量的担忧,FDA近年来一直与USP积极合作,希望USP及相关的利益方能够对OTC药品各论的质量标准进行更新,以符合FDA对其质量要求。
- 仿制药:仿制药杂质研究一般参考相应的法规指南,制定的杂质标准基本原理如下:
- 与原研药进行比较,使用相同的验证、稳定性指示分析方法;
- 证明所述杂质是药物的重要(人类)代谢物,假设代谢物的水平与质量或疗效无关;
- 使用适当的科学文献证明杂质水平是安全的;
- 通过强制降解,分析可能的降解产物;
- 通过处方工艺研究和稳定性研究,分析可能的杂质;
- 用毒性研究评价杂质的水平;
- 收载在药典各论中的杂质限度可以作为仿制药标准的参考。
随着质量源于设计(QbD)越来越成为行业的标准,对杂质来源的全面了解将成为药物整个开发过程的一部分。在仿制药开发完整的质量目标产品概要(QTPP)时,需要考虑药物杂质及其来源。ICH Q8指南将QTPP定义为药物产品质量特性的前瞻性总结。理想情况下,该质量特性将用于确保药品的预期质量。随着QTPP的开发,申请人将把所需的产品与需要满足的关键质量属性(CQA)关联起来,以确保产品质量、安全和性能。
制剂中降解产物可归类为CQA,在药物开发过程中根据对产品的安全性和有效性影响进行评估。药物辅料的关键材料属性(CMA)包括影响药物产品的杂质,这些CMA应在初始开发期间以及在实施批准前进行研究。
结束语
本文讨论了制剂杂质的研究思路。杂质研究关系着药品的质量和有效性,因此它贯穿药品研究的始终。制剂杂质研究不同于原料药中杂质研究,降解杂质是制剂杂质研究的重点,降解条件和分析方法的选择决定了杂质的产生和检出。将制剂中的杂质控制到安全、合理的范围内关系到药品从研发、生产,到上市后的整个生命周期。
<END>



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论