

































随着新药研发技术的不断进步和审评制度改革,创新药投入临床使用的速度正在加快,这是另所有研发人员振奋鼓舞的事情,但是一个全新化合物如此快速投入临床并应用于人,相应的风险也随之增加,这个风险一方面来源于化合物本身,一方面来源于杂质,因此对杂质进行科学系统的研究,对于防范和降低临床风险意义重大。
1.杂质定义
什么是杂质呢?杂质是指存在于原料药中的与原料药结构不一致的任何成分。药物在生产、储存和运输过程中都有可能产生杂质。
2.杂质分类
杂质的分类方式有很多,按照理化性质可以分为无机杂质、有机杂质和残留溶剂;按照产生途径可以分为工艺杂质和降解杂质;按照其毒性可以分为普通毒性和毒性杂质;按照其控制策略可以分为特定杂质和非特定杂质;按照其产生的风险可以分为潜在杂质和实际存在杂质。
对杂质的分类可以根据分析目的进行选择,例如工艺研究总结中通常需要对杂质产生途径(降解杂质、工艺杂质)进行描述;在安全评价中则需要区分是普通杂质还是毒性杂质,在分析方法选择时通常是按照有机杂质(含DNA反应性杂质)、无机杂质、残留溶剂来分类。
3.杂质研究的基本思路
2010年杂质谱控制的理念被提出,使得杂质研究由个别杂质控制上升为系统控制,杂质研究的工作也不再是单纯的杂质检测与分析工作,成为了一项需要由工艺、质量、制剂、安全性评价等多学科多领域共同完成的系统工程。
杂质谱研究的思路和策略源于QbD理念,经过多年的摸索和研究,已经从以终为始的被动控制模式转化为以源为始的主动控制模式,也就是从杂质的来源入手,结合产品的结构特点、生产工艺、处方组成、存储条件、稳定性特征等信息全面分析产品中可能存在的杂质,然后对杂质存在的可能性大小,是否容易清除、安全等级进行评估,建立适宜的分析方法,确保有效检出和确认各种潜在杂质;在此基础之上,结合生产工艺、稳定性特征,跟踪杂质在安全性或临床试验结果中产生的影响,在符合指导原则的前提下制定每个特定杂质的限度;最后是对杂质的来源和去向进行分析,通过工艺控制、包装改进或储存条件调整来有效抑制杂质的产生,从源头上有效把控杂质。
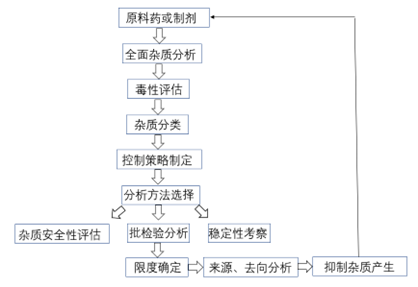
图1 创新药杂质研究一般思路
本文按照无机杂质、残留溶剂和有机杂质的顺序逐一探讨各类杂质的具体研究思路和控制策略。
3.1无机杂质
药品中的无机杂质包括残留的催化剂、配体、试剂、重金属及无机盐等。对于无机盐和普通试剂可以参照药典附录中的方法进行控制,例如氯化物、硫酸盐、炽灼残渣等检查项。在无机杂质中重点应关注的是元素杂质。
元素杂质的来源包括有意添加的无机试剂或催化剂引入;原辅料中潜在的元素杂质;生产设备引入及包装系统中浸出的元素杂质,依据风险评估,元素杂质可以分为4个风险等级(I、2A、2B、3),其中I及2A类元素杂质无论是否有意添加均应进行风险评估,2B类元素杂质仅在有意添加时进行风险评估,3类元素杂质,有意添加时应进行风险评估,在非有意添加时依据所选择的剂型决定是否进行风险评估。元素杂质的控制策略中,有一个“控制阈值”值得关注,控制阈值是元素水平相当于PDE的显著性水平,相当于PDE 30%,当多批既定工艺条件下的样品检测结果表明所有来源的某种元素杂质的残留水平均低于控制阈值,那么,在对数据进行评估并证明已对该元素杂质进行了足够控制的情况下,无需额外控制。否则,需建立控制方法加以控制。举个例子,比如某口服制剂原料药的起始物料的生产工艺中使用了金属催化剂钯(PDE 100μg/天,元素允许浓度10μg/g),原则上需要考虑钯所带来的风险,当我们检验了多批起始物料、中间体及成品后发现钯的残留量均为未超过3 μg/g(PDE的30%),那么我们无需建立控制方法。
3.2残留溶剂
残留溶剂指原辅料生产或制剂制备过程中使用或产生的有机挥发物。依据风险评估,残留溶剂可以分为4类,1类溶剂具有不可接受的毒性或危害环境,应避免使用;2类溶剂具有非遗传毒性的动物致癌性,可能导致神经毒性、致畸性等不可逆毒性或其他严重但可逆的毒性,应限制使用;3类溶剂属于低毒性溶剂,限度可按照5000ppm或以上来控制。4类溶剂属于没有足够毒理学数据的溶剂。其中,前3类溶剂在ICH指导原则中均有明确的限度,第4类溶剂由于缺乏足够的毒理学数据没有明确的限度。在对第4类溶剂进行研究时,应对毒性数据库和相关文献进行全面检索,将所能获得的毒性数据汇总,分别计算限度(计算方法参考ICH Q3C),然后选择其中的最低限度作为该溶剂的限度,当有新的研究数据发表并且所计算的限度更低时应及时对限度进行修订。
在对残留溶剂进行分析时,除了实际使用的溶剂还应注意潜在可能产生的溶剂,如同时使用甲醇和乙酸可能生成乙酸甲酯、使用甲苯时需同时控制其中可能存在的苯等。
3.3有机杂质
有机杂质主要是指有关物质,也就是与生产工艺和药物结构有关的杂质。有关物质通常包括:起始物料、中间体、副产物、降解产物等。有关物质的控制是药物质量控制的重点内容,控制的难度也较无机杂质和残留溶剂要高。
按照杂质谱研究的一般规律,有关物质研究首先也是对原料药或制剂产品中可能存在的杂质进行全面的分析,列出所有的潜在杂质并对杂质产生的可能性大小和毒性进行评估和分类,1)无论理论上产生可能性高低,实际未检出或检出量较低,且稳定性中未见明显增长的杂质,可以按照非特定杂质来控制。2)对于那些实际检出量很高或稳定性中明显增长的杂质,应按照特定杂质来控制。3)对那些含有警示结构的杂质应先按照DNA反应性杂质进行评估,如在充分研究后排除了其致癌致突变性,则可按照一般杂质来控制,否则列入DNA反应性杂质控制队伍之中。
在确定初步的研究策略后我们再来看下各种杂质限度如何制定。
3.3.1 普通杂质
ICH Q3A和Q3B中分别给出了原料药和制剂产品中杂质限度汇总表。
表1 原料药杂质限度

表2 制剂杂质限度
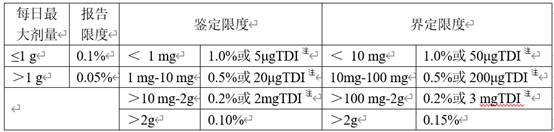
注:取限度低者
对于创新药,非特定杂质(即单一最大杂质)可按照上表中的限度进行控制,而特定杂质在有充足数据支撑的前提下可以超越指导原则中的限度要求,这是因为创新药在申请临床研究之前开展大量的毒理学研究,那些含量超过鉴定限或界定限的杂质以及稳定性性研究中明显增长至超过鉴定限或界定限的杂质都可以在毒理学研究中得到安全性评价,所获得数据可以用于支持相应杂质限度的制定。
举个例子:
1)某原料药中杂质A含量为0.13%,该批原料药用于犬长毒实验,NOAEL值为40 mg/kg/天。假设临床剂量为150mg.d-1,以患者体重60kg计,则人的给药剂量为2.5mg.kg-1.d-1,与犬的给药剂量比较,可支持该杂质的安全限度应为(40/2.5)*0.13%=2.08%
2)6批一定规模样品中杂质A平均含量0.13%,标准偏差0.04%,该杂质限度0.25%(平均值+3 SD)。
3)3批上述原料药长期放样24个月,杂质A含量最高增长至0.56%,依据稳定性结果该杂质限度为0.6%。
综上,依据安全性、工艺可行性及稳定性的结果综合制定该杂质的限度可定为0.60%。
3.3.2 DNA反应性杂质
在有关物质中还有一类特殊的杂质——DNA反应性杂质。这一类杂质即使在较低水平也能引起DNA损伤。对于这一类杂质,应严格控制其含量。依据ICH M7致癌性及致突变杂质可以分为以下5类。
表3 根据致突变潜力和致癌潜力对杂质进行分类及控制措施
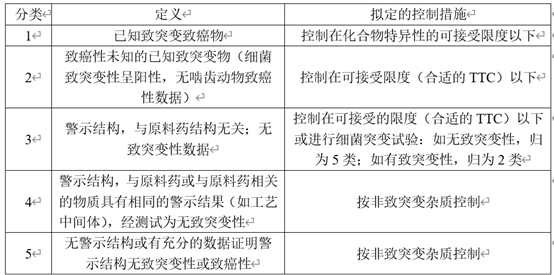
在对杂质进行分析时,首先检索文献和数据库注,如果有充分数据证明警示结构无致突变性或致癌性则归为5类,有致突变性,但致癌性未知的杂质归为2类,同时具有致突变和致癌性的杂质归为1类,当没有致突变的相关数据时,则应开展细菌突变试验试验或动物试验来对杂质的致突变性进行预测或检测,将杂质归为3类、4类或5类。
注:如果有Q-SAR预测软件(基于统计学和专家规则各一个),可以用软件预测替代文献或数据库检索以及细菌或动物试验,这样可以提高工作效率、降低成本。
当杂质被评估具有致突变或致癌性时,限度要怎样确定呢?有以下四种计算方法:
1)基于TTC的可接受摄入量
一个致突变杂质每天摄入1.5 μg所引起的风险被认为是可以忽略的,这一数值可以适用于大部分药物。
2)基于杂质特异性风险评估计算可接受摄入量
①具有阳性致癌性数据的致突变杂质
----可以采用线性外推法:通常以啮齿类动物的TD50除以50000作为杂质的可接受摄入量,相应的患癌风险为1/100000。
----采用结构相似杂质的可接受限度:如果没有查到化合物的致癌性数据,但是可以查到与待研究杂质化学结构类似的已知致癌物的致癌性数据,可以采用化合物特异性的方法来计算待研究化合物的可接受摄入量。
②有实际阈值的致突变杂质
可以通过NOAEL值和不确定因子计算PDE,具体计算方法可以参见ICH Q3C。
3)与短于终生(LTL)暴露相关的可接受摄入量
假设致癌性风险随暴露量(给药量)增加而增加,那么终生以低剂量持续给药的患癌风险与累计暴露量相同的短时间给药的患癌风险等同。因此,短时间给药的情况下TTC的值可以高于1.5 μg/天,这种LTL的概念可适用于临床研发阶段(要求临床研究时间不低于1月)以及上市后有明确预期治疗期限的药物。
4)多个致突变杂质的可接受摄入量
当药品中含有2个2类或3类杂质,应制定各自的限度;当含有大于等于3个2类或3类杂质,应总致突变杂质限度。限度设置可参考下表设置。
表4 多个致突变杂质可接受摄入量

DNA反应性杂质的控制方法有4种:
方法1:终点控制,即将杂质列入终产品质量标准,如果连续3批生产批或连续6批中试批样品中致突变杂质水平低于限度30%,则可以采用定期确认行检测的方式控制,否则应将该杂质的检测作为常规检测项。
方法2:在起始物料或中间体设置中间控制点,将杂质水平控制在可接受限度内。
方法3:在起始物料或中间体设置中间控制点,杂质控制标准可高于可接受限度,结合工艺控制和对杂质清除和去向的理解确保终产品杂质的水平低于可接受限度。
方法4:基于对工艺和杂质特性的理解,经过科学论述,确信终产品中杂质水平低于可接受限度,则无需对该杂质进行检测。
4.小结
杂质研究是贯穿于新药研发始终的重要内容,是药品安全性的重要保障,近年来提倡的杂质谱研究更是将杂质研究上升为科学系统的研究工程,在创新药的研发中应正确认识杂质谱研究的重要性,在对杂质进行全面分析的基础上结合安全性评价结果、稳定性特征、规模放大时杂质谱的变化,进行风险评估,制定科学合理的控制策略,为临床研究及上市后药品的安全性提供保障。
相关阅读:
《药物基质亚硝胺(NDSRIs)杂质管理:安全性与可操作性的博弈》
参考文献
[1]ICH指导原则Q3A、Q3B、Q3C、Q3D、Q6A、Q11等。
[2] ICH 指导原则M7
[3]化学药物研究技术指导原则
[4]仿制药杂质研究与控制的基本思路与策略 张哲峰
[5]浅谈杂质限度制定的一般原则 张哲峰
-END-




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论