

































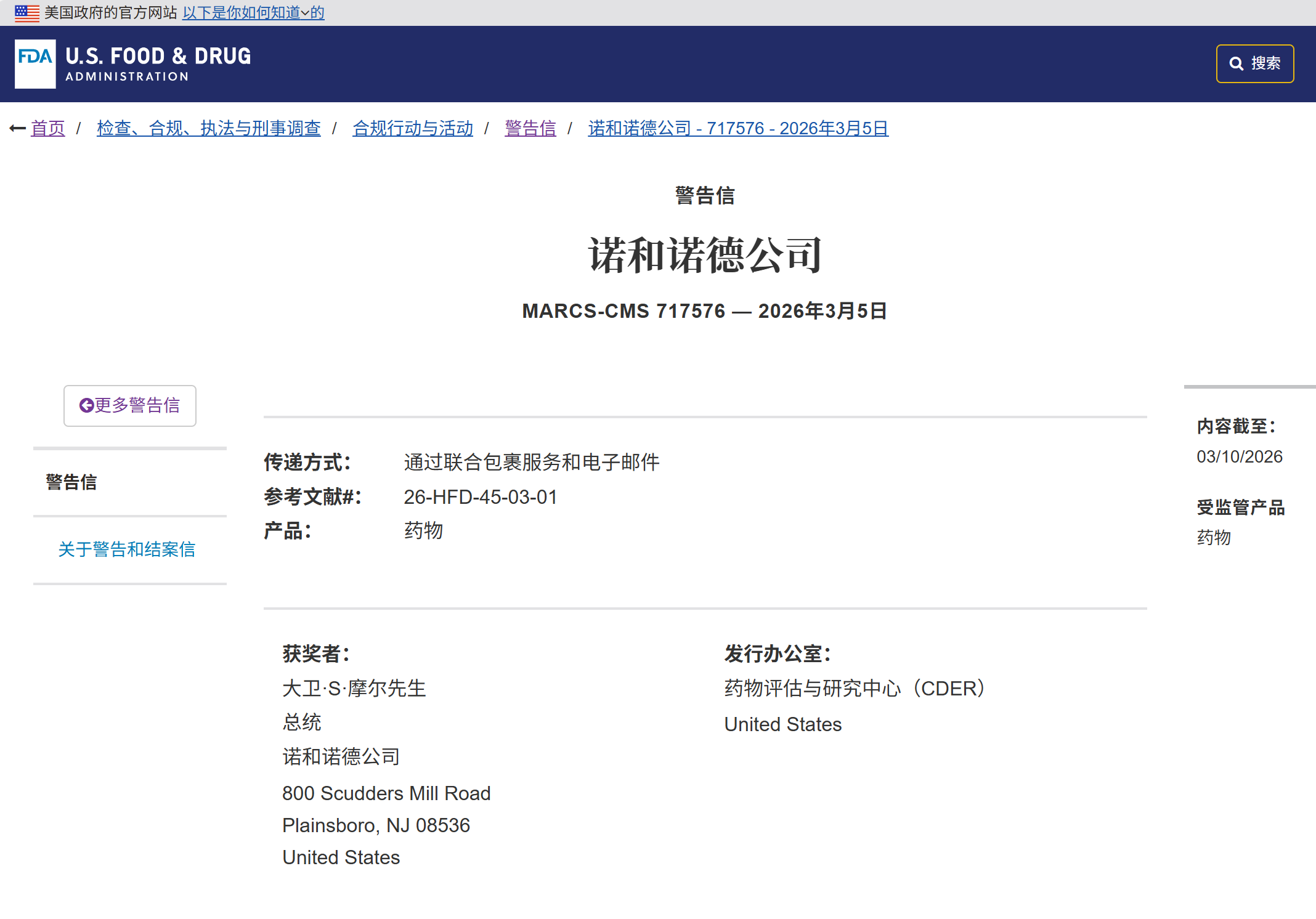
2026年3月5日,美国FDA向诺和诺德(Novo Nordisk)发出严厉警告信。
此次违规并非源于司美格鲁肽的生产质量(cGMP)缺陷或临床数据造假,而是触犯了制药合规中最敏感的红线——上市后不良体验(PADE)报告的系统性失效。
此前,其位于印第安纳州布卢明顿的工厂因严重的无菌控制问题被判定为“不可接受状态”。
从药物警戒(PV)的逻辑崩塌到核心供应链的底线失守,这家GLP-1巨头的合规隐患在高速扩张中彻底暴露。
致命误判:PV体系的“因果论”陷阱
FDA警告信直指诺和诺德内部程序(SOP Q014048)的致命漏洞:公司允许员工或外包商在认为事件与产品“无关”时,直接拒绝接收或取消报告。
这一“先评估、后报告”的逻辑,直接违反了21 CFR 314.80的核心要求——只要满足四要素(可识别患者、报告人、产品及事件),无论因果关系如何,均必须上报。
严重漏报:涉及利拉鲁肽的中风致残案、司美格鲁肽的死亡病例,仅因内部人员主观判定“感觉无关”或“信息不全”,便被拦截在报告系统之外。
流程延误:一起严重自杀意念案件在“医学审查”环节滞留近两个月,远超法规时限。
管理失控:大量本应跟进的严重事件被错误归类为“无效”;过度依赖外包呼叫中心却缺乏监管,导致含关键信息的录音被丢弃;IT系统缺陷更致使案件长期处于“待审查”假死状态,风险预警全面失效。
并购包袱:无菌生产的“猫毛”危机
2024年12月,诺和诺德完成对Catalent布卢明顿工厂的收购,旨在解决产能瓶颈,却意外继承了一个长期存在质量积弊的“问题工厂”。
2025年6月至7月,FDA检查发现该无菌生产环境中存在猫毛和人类毛发等异物,且微生物控制程序缺失。
尽管诺和诺德试图整改,但FDA评级迅速升级:
2025年7月:开具Form 483,列出多项严重缺陷。2025年10月:定级为OAI(Official Action Indicated),意味着工厂处于“不可接受的合规状态”。
2025年12月11日:正式发出警告信,指出根本原因分析不充分,受污染药品仍在释放。
连锁反应:产业链的“蝴蝶效应”
布卢明顿工厂的危机引发了严重的行业震荡。生物科技公司Scholar Rock治疗脊髓性肌萎缩症(SMA)的重磅药物apitegromab因在该厂生产,于2025年9月被FDA拒绝批准。
巨头再生元(Regeneron)的核心眼科药Eylea HD审批也被迫延期,新设施获批需耗时9至12个月,短期难解困境。
行业警示
诺和诺德的教训惨痛而深刻。它揭示了制药行业在并购扩张时的隐忧:“收购”不是免责金牌,“外包”更非监管盲区。
在GLP-1赛道竞争白热化的当下,安全监测能力与生产合规水平(特别是数据完整性、无菌保障及交叉污染控制)已成为比产能更关键的生存底线。
诺和诺德这一跤,摔醒了整个行业:在FDA的聚光灯下,没有“意外”的漏报,也没有“无辜”的收购,只有“必然”的惩罚。
文章来源:蒲公英Ouryao
原文链接:https://mp.weixin.qq.com/s/C8Jnt_Xrdcg71j5ZZSsKIw
扩展阅读:




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论