

































在创新药与改良型新药的研发管线中,以及仿制药的深度评价过程中,生物等效性(Bioequivalence, BE)试验始终扮演着决定产品能否顺利迈向商业化与全球市场拓展的核心角色。通过严谨地比较受试药品与参比药品的等同性,研发团队能够科学地推断两者在临床治疗效果上的一致性。
在当前的全球监管体系下,无论是在评估企业基本面还是推进产品合规上市时,美国FDA等权威监管机构对评价指标设定了明确的优先层级:首选药代动力学(Pharmacokinetic, PK)终点指标 ;其次为药效动力学(Pharmacodynamic, PD)终点指标 ;再次为临床终点指标 ;最后为体外终点指标。
在绝大多数常规研发项目中,PK终点是评价BE的“金标准”。通常采用统计学中的置信区间(Confidence Interval, CI)法,当主要PK参数经对数转换后,几何均值比(GMR)的90% CI落在80.00%~125.00%的等效性区间内,即可认定两制剂吸收的速度和程度相当。
然而,生物医药的管线布局往往充满挑战。部分特定作用机制的药物(如局部作用药物,或进入体循环量极低、浓度难以测定的全身药物),传统的PK评价体系面临“无液可采、无靶可测”的窘境。此时,转向以药效学(PD)指标作为替代终点,不仅是监管合规的有效路径,更是研发策略的优选。相较于耗资巨大、周期漫长的临床终点研究,药效学研究具备易于开展、重复性高且研发成本更为可控的显著优势。
本文将深入剖析几大特殊给药途径及特殊药物的药效学BE评价策略,为药企的研发规划与合规策略提供深度参考。
一、 消化道局部作用药物的破局思路
对于以胃肠道为直接作用靶点、几乎不产生系统性暴露的药物,其BE评价的复杂性远超常规系统分布药物。研发设计必须将药物在靶部位的递送效率、理化特性与具体作用机制紧密结合。
(一) 阿卡波糖(Acarbose):精准捕捉血糖降幅的动态变化
阿卡波糖作为一种经典的口服 α-葡萄糖苷酶抑制剂,其核心机制在于通过抑制小肠壁细胞的酶活性,延缓多糖、双糖的降解吸收,从而控制餐后血糖。由于其作用靶点明确锁定在肠道局部,且极少吸收入血,常规血药浓度检测失去了评价意义。
阿卡波糖化学结构
基于此,美国FDA在相关指导原则中明确推荐以血清血糖的变化作为药效动力学效应指标。在试验设计上,FDA建议采用随机双交叉设计,并设置1周的清洗期。极其关键的一环在于正式试验前的预试验:这不仅是为了探索能产生降糖药效的最低剂量(需避开量-效曲线的坪剂量区域),更是为了精准测算正式试验所需的受试者样本量。
在数据采集中,基线校正是核心逻辑。受试者需在给药前一天服用标准化蔗糖水(75g蔗糖溶于150mL水),并密集采集0~4小时的血液样本建立基线;次日则将阿卡波糖与蔗糖水同服,在相同时间点采血。
评价指标方面,FDA聚焦于两点:
① ΔCmax:即给药前一天(单服糖水)与给药当天(药糖同服)血糖浓度峰值间的最大差值。
② AUEC0-4h:即0~4小时血糖浓度-时间曲线下面积的差值。当受试药品与参比药品的 ΔCmax 和 AUEC0-4h 均值比的90% CI均落在80.00%~125.00%时,即可判定等效。
值得注意的是,针对不同人群的种族差异,评价体系需要灵活调整。国内多项研究指出,在中国人群中开展阿卡波糖BE试验时,由于潜在的体内生物转化差异,并不推荐直接使用FDA推荐的 AUEC0-4h。
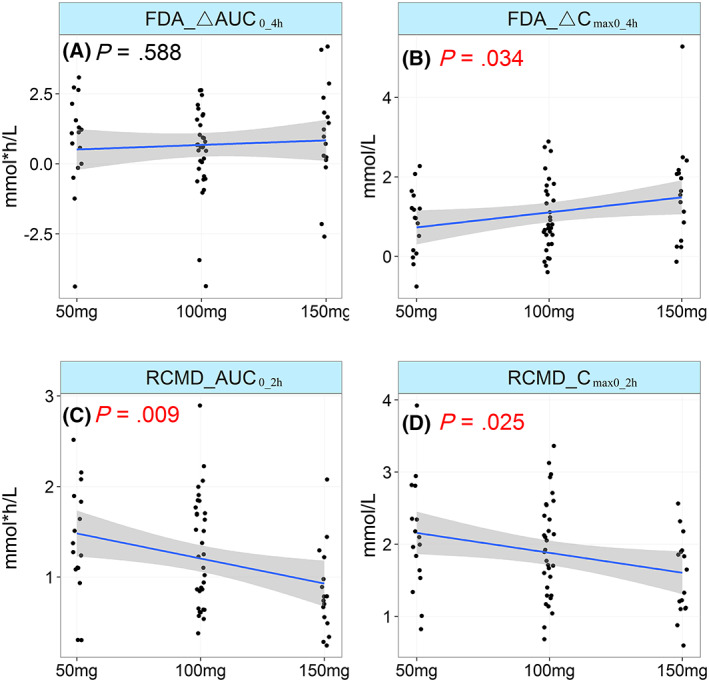
FDA推荐的ΔAUC0–4h在不同剂量的阿卡波糖之间无显著差异(P = 0.588)
中国药科大学的探索性研究提出,建议采用服糖与药糖同服的最大血糖浓度差值(ΔCSG,max)、血糖曲线最大值与最小值差值(GE及GE')、平均稳态血糖浓度(Css)以及波动面积(fAUC)这5个参数作为更适宜中国人群的等效性评价指标。而拜耳公司在德国的三周期交叉设计研究中,则采用了最大血糖浓度比值(Ratio Cmax)和0~4小时曲线下面积比值(Ratio AUC)作为主要指标。这提示我们在跨国临床推进与全球申报时,需高度重视靶点人群的区域性代谢特征。
(二) 奥利司他(Orlistat):从粪便排泄率洞察抑制效能
作为胃肠道胰脂酶抑制剂,奥利司他通过阻断脂肪水解来减少热量吸收。由于同样几乎不吸收入血,FDA在2010年发布的草案中,创新性地将“稳定状态下24小时粪便中排出的脂肪量与每日摄入脂肪量的比率”定为药效学终点。
奥利司他化学结构
该试验要求进行三交叉多剂量给药,严格要求受试者摄入标准化饮食(脂肪供能占比严格控制在30%)。通过长达9天的试验周期与至少4天的洗脱期,结合最大效应(Emax)模型的剂量标度法计算相对生物利用度,最终依据90% CI是否落在80%~125%来判定等效性。
二、 皮肤外用制剂:攻克透皮吸收的评估壁垒
皮肤外用药物通过顺次渗透角质层(约 10μm)、表皮层(约 100μm)至真皮层(约 1000μm)发挥局部疗效(如抗炎、抗菌、麻醉等)。因其系统暴露量极低,基于血液样本的传统PK研究通常折戟沉沙。目前,学术界与工业界主要通过药效学研究、临床终点研究或体外-体内相关性(IVIVC)研究来破局。
(一) 皮质类固醇激素:血管收缩测定法(VCA)
对于靶向表皮/真皮层起效的皮质类固醇药物,血管收缩测定法(VCA,亦称人体苍白斑试验,HSBA)是目前FDA认可的体内药效学方法。其生理学机制在于药物渗透后引起局部微血管收缩,产生肉眼可见或可通过比色计精准量化的苍白反应,且苍白程度与临床抗炎疗效高度相关。
在研究设计上,采用复杂的药效动力学群体模型(Emax 模型)。其核心数学表达为:
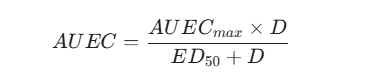
其中,AUEC 为药效-时间曲线下面积,D 为给药持续时间,ED50 为达到半数最大效应所需的给药时长。
实操中,极为考验临床试验运营能力的是 ED50 的精准估算。预试验需利用参比药品(RLD)建立剂量持续时间-效应曲线,并确定两个关键给药时长:D1(约 0.5× ED50)和 D2(约 2 × ED50)。在正式试验中,只有当受试者在 D2 与 D1 持续时间下的效应比值(AUECD2/AUECD1)大于1.25时,其数据才被认为具备足够的灵敏度并纳入最终的BE评价体系。需要注意的是,VCA的适用范围仅限皮质激素类药物,无法外推至其他靶点的外用制剂。
(二) 新兴取样技术的突围
针对非激素类外用药,行业正在积极验证新技术:
1. 胶带粘贴法(DPK):通过连续剥离角质层来获取药物浓度,被认为是评价角质层为靶位(如抗真菌药)的有效手段。但其局限在于无法评估作用于表皮-真皮深层的制剂,且不同实验室间的数据重现性仍是一大考验。
2. 皮肤微透析(DMD)技术:一种微创的在体取样技术,通过在真皮层植入探针,动态收集透析液以检测游离药物浓度。它是获取深层组织暴露量数据的理想方案,但对于高脂溶性或易与组织蛋白结合的分子,探针回收率低下仍是亟待突破的工艺瓶颈。
3. 开放流动微灌注(dOFM):为了跨越这一技术鸿沟,真皮开放流动微灌注(dermal Open Flow Microperfusion, dOFM)技术应运而生并迅速获得了监管层的高度关注 。dOFM彻底摒弃了半透膜设计,采用具有宏观开孔的探针结构 。在灌流过程中,流体通道与周围的组织间液完全贯通,药物分子的采集不再依赖于微观扩散梯度,而是通过直接的流体力学“对流”将未经物理过滤的组织液抽取出来 。
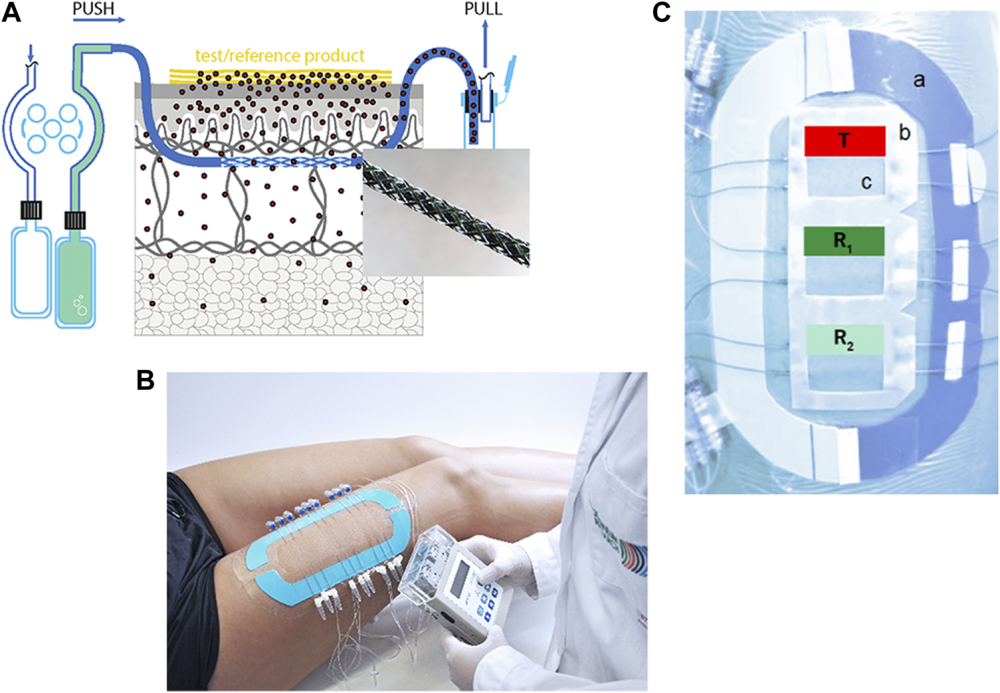
(A):dOFM采样装置的示意图,并放大了交换区域。(B):完整的临床装置,包括三个应用部位、九个插入的dOFM探头和一个可穿戴泵。(C):使用稳定环(a)来最大限度地减少皮肤变形。标准化的应用框架(b)精确地定义了应用部位(c)。T(受试药物),R1,2 (参比药物)。
三、 吸入与喷鼻制剂(OIPs):解构双通道吸收的复杂性
局部作用的喷鼻剂与口腔吸入制剂的BE评价被公认为仿制药开发领域的“深水区”。其核心痛点在于:药物进入呼吸道后,既有实现局部疗效的靶部位沉积(Lung/Nasal deposition),又伴随着经由黏膜或吞咽入胃肠道导致的系统性吸收。疗效与血药浓度常常出现非线性脱节。
(一) 喷鼻剂的阶梯评价策略
根据FDA指南,喷鼻剂的评价依据剂型呈现两极分化:
- 溶液喷鼻剂:由于主药已完全溶解,监管部门认为只要处方一致(Q1/Q2相同)、包装密闭系统相似,仅凭严格的体外测试(如单次喷出量、激光衍射测定雾滴大小、喷雾模式与几何学等)即可充分证明生物等效。
- 混悬喷鼻剂:因难溶性微粒的粒径分布直接影响局部溶解吸收,且尚无单一的体外模型能完美模拟这一过程,监管要求极为严苛。除7项体外研究外,必须附加体内验证。若能检测血药浓度,则采用“体外+局部临床+PK等效”组合拳;若无法检测,则必须转向“体外+局部临床+药效学(或全身临床)”评价矩阵。
(二) 口腔吸入制剂:活性炭阻隔与药效学终点
对于哮喘和COPD等适应症的经口吸入制剂,EMA设置了极为严苛的“纯体外评价”豁免条件(需满足包含装置操作、气流阻力、空气动力学表现等9大前提标准)。若任一不符,则必须开展体内等效性评价。
评价吸入制剂需双管齐下:基于全身暴露量的安全性评价,以及基于肺部沉积量的有效性评价。
在肺部有效性评价中,活性炭阻隔法是一种精妙的PK辅助设计。以特布他林气雾剂为例,受试者在吸入给药前后口服活性炭混悬液,利用活性炭强大的物理吸附能力,彻底阻隔并清除经吞咽进入胃肠道的药物。此时从血浆或尿液中测得的PK参数(如 AUC 和 Cmax),便能纯粹且精准地反映出药物在肺部的真实沉积量,进而用于验证试验药物与参比药物的有效性等效(90% CI需落于80.00%~125.00%)。中国NMPA出台的指导原则亦强调了标准化吸气流速培训及吞咽控制在试验中的重要性。此外,二维闪烁成像技术亦可作为直观测定不同肺区段放射性沉积比例的替代手段。
若PK数据难以获取,药效学研究(单剂量支气管扩张或支气管激发试验)则是不可或缺的核心证据。
- SABA(短效 β2 激动剂)与 LABA(长效 β2 激动剂):研究主要在哮喘患者中开展,考察气道功能的逆转能力。支气管扩张试验的黄金指标为测量单剂量给药后 FEV1(1秒钟用力呼气量)的变化量及曲线下面积(FEV1 AUC)。支气管保护研究则运用乙酰甲胆碱等激发剂,测定诱发 FEV1 下降20%所需的激发浓度(PC20FEV1)或剂量(PD20FEV1)。FDA针对沙美特罗等特定产品的指导原则详细规定了受试者入组门槛(如基线 FEV1 预计值为40%~85%,需具备可逆性)以及详尽的洗脱及导入期管理规范。
- 吸入性糖皮质激素(ICS):通常要求进行更长周期的临床终点研究,以证明受试制剂非劣效于参比产品,主要疗效终点依然是肺功能指标。
四、 特殊全身起效药物的指纹溯源与药效学替代
不仅局部用药面临传统PK评估的困境,某些全身起效的大分子或复杂混合物,因体内代谢特性的特殊性,同样无法单纯依赖血药浓度曲线来判定等效性。
(一) 低分子量肝素(LMWH):双靶点酶活性的精细刻画
低分子量肝素作为抗凝血的基石药物,其本质是一系列不同聚合度、不同分子量的多糖链混合物。受限于当下的分析科学技术,极难对大于3600Da的多聚糖链进行完整的序列测定与体内代谢追踪。
因此,监管机构(包括FDA、EMA及中国NMPA)在审评此类产品时,建立了一套严格的多维评价体系:在确认理化性质、原料解聚方式、寡糖序列特征及体外生物学活性等同的基础上,强制要求开展人体药效学等效研究。
其药效学终点精准锚定于凝血级联反应的关键节点:血浆抗FXa活性与抗FIIa活性。试验要求采用单剂量交叉设计,核心评价参数包括峰值效应(anti-Xamax 与 anti-IIamax)、效应-时间曲线下面积(AUEC0-t)、达峰时间与半衰期。受试与参比药物在 AUEC 与 Emax 上的GMR的90% CI必须落入80.00%~125.00%区间。由于人体内源性FXa/FIIa基线水平会受昼夜节律及生理状态的剧烈波动影响,这一评价过程对临床检测方法的灵敏度与基线校正的数理模型提出了极高的挑战。
(二) 干扰素(Interferons):寻找下游信号的生物标志物
干扰素在抗病毒、抗肿瘤及免疫调节管线中占据重要地位。然而,其给药后在血浆中的游离浓度极低,常低于常规酶联免疫或细胞病变抑制法的检测下限(LLOQ)。
面对这一难题,研发人员将目光转向了干扰素介导的下游信号通路。当人体接受干扰素刺激后,会强烈诱导产生抗病毒蛋白——2',5'-寡聚腺苷酸合成酶(2',5'-OAS)。2',5'-OAS既是干扰素发挥抗病毒作用的核心效应物质,又是其体内药理活性的“放大器”。临床研究证实,诱生出的2',5'-OAS曲线下面积与注射的干扰素剂量呈现出高度相关的量效关系。因此,将2',5'-OAS作为药效学替代终点,成为评价干扰素类药物体内生物等效性的一把“金钥匙”。
五、结语
在现代生物制药工业中,依靠单一的药代动力学来包打天下已不符合日益复杂的给药系统与药物机理特征。以药效学(PD)为终点的BE评价体系,实质上是对药物在体内真实生物学效能的深层次还原。
从消化道的降糖控脂代谢干预,到皮肤深层的靶向递送;从呼吸道双重吸收通道的精妙分离,到复杂生物混合物的靶点酶活性追踪,每一个药效学指标的选定,无不依赖于前期扎实的非临床机理探索与数理统计模型的严密构建。对于深耕创新药与高端仿制药管线的医药企业而言,深刻理解FDA、EMA以及NMPA的前沿指导原则,并在试验设计初期与审评中心进行充分、高频的沟通与探讨,将是打破研发同质化内卷、确保产品全生命周期合规与商业变现的关键所在。
扩展阅读:
1. 解锁药物开发密码:早期药物盐与共晶的系统化筛选、评价与放大生产及制剂转化全解析
3. 深度解析生物利用度:从Cmax/AUC到FDA监管,揭秘制剂工艺与临床疗效的核心纽带
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论