


































在现代药代动力学(Pharmacokinetics, PK)与生物药剂学的交叉领域中,生物利用度(Bioavailability, BA)始终占据着核心的学术与临床地位。生物利用度被严格定义为:药物的活性物质从其制剂中释放,并经吸收进入体循环,最终到达药物作用靶部位的速率与程度。在临床药理学实践中,这通常通过血浆药物浓度-时间曲线来进行量化评估。
同一化学结构式的药物,若被加工成不同的剂型(如片剂、胶囊、溶液),其生物利用度往往存在显著差异。即便剂型相同,不同制药企业的工艺水平差异,乃至于同一企业不同批次的产品,也可能在BA上表现出统计学上的不一致。这种差异在临床上具有深远的影响:对于起效时间要求严苛的药物(如镇痛药、催眠药),吸收速率(主要由峰浓度 Cmax和达峰时间 Tmax决定)的微小波动可能导致起效延迟;而对于需要维持稳态血药浓度以治疗慢性疾病的药物,吸收程度(主要由药时曲线下面积 AUC 决定)的不足则会导致治疗失败。因此,全面、系统地评价药物制剂的生物利用度,不仅是指导新药处方开发与工艺优化的“金标准”,更是探寻临床药物无效或毒性反应根源的关键科学路径。
一、核心测度——生物利用度的评价方法体系
根据监管科学的优先次序,生物利用度的研究方法可分为体内(In vivo)与体外(In vitro)两大类。申请人应基于药物的理化性质与分析条件,选择最具敏感性、准确性与重现性的评价策略。
1. 体内评价方法:血药浓度法(The Gold Standard)
血药浓度测定法是目前国际公认的评估人体生物利用度最为普适且可靠的方法。该方法的核心逻辑在于:对于绝大多数全身起效的药物,其在中心室(如全血、血浆或血清)中的浓度变化曲线,能够按比例反映其在靶组织受体部位的暴露量,并与最终的药理学或毒理学效应存在高度相关性。
在数据处理层面,非房室模型分析法(Non-compartmental Analysis, NCA)因其较少的理论假设而被广泛采用。通过在给药后密集采集血液样本,可计算出三个决定BA的关键参数:
- 药时曲线下面积(AUC):反映药物吸收入血的总量(吸收程度)。通常采用线性/对数梯形法计算至最后一次可测浓度的 AUC0-t,并外推计算出AUC0-∞。
- 峰浓度(Cmax):药物在体内的最高浓度,是吸收与消除过程达到动态平衡的转折点,直接反映吸收速率与潜在的毒性阈值。
- 达峰时间(Tmax):反映药物吸收的速度。
从统计学角度考量,个体的AUC与 Cmax通常呈现对数正态分布。根据药代动力学基本公式:
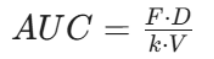
其中F为生物利用度,D为剂量,k为消除速率常数,V为表观分布容积,通过自然对数转换,可以将原本的乘除关系转化为线性加减关系:ln(AUC) = ln(F) + ln(D) - ln(k) - ln(V)。这一数学转换消除了个体间 k 和 V 的乘积性影响,使得后续的方差分析(ANOVA)满足正态性与方差齐性的统计学前提。
2. 体内评价方法:尿药浓度法(The Alternative Pathway)
尽管血药浓度法占据主导地位,但在特定场景下,如:药物缺乏高灵敏度的血样定量分析方法、药物表观分布容积过大导致血药浓度极低、或医疗采血条件受限时,尿药排泄数据处理方法便成为一种极具价值的替代方案。
尿药浓度法的理论适用前提非常严苛:药物必须有很大比例(通常要求>70%)以原形分子从肾脏排泄,且其肾排泄过程需符合一级动力学过程。这意味着,尿中原形药物的出现速度(R)必须严格与当时的体内药物总量成正比。通过收集给药后至少7个消除半衰期(t1/2)以上的完整尿液,可计算出累积排泄量(Ae)。
尿药累积曲线
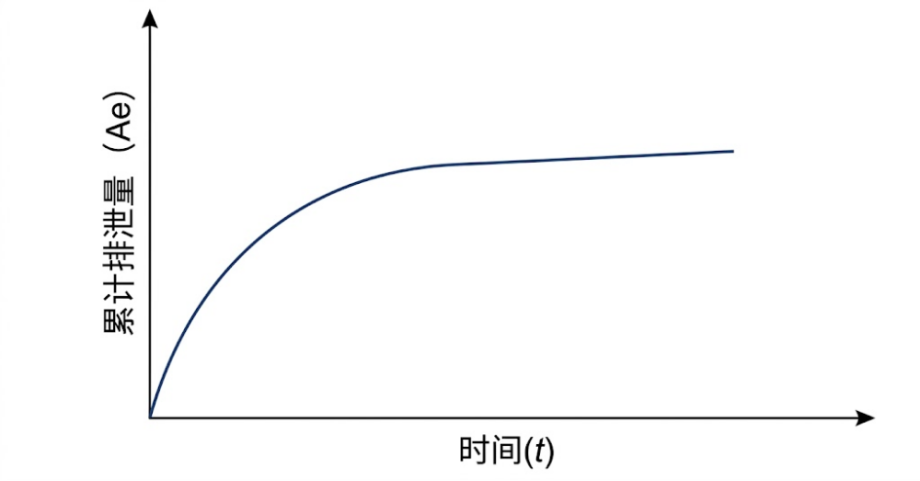
随着时间的推移,累积排泄量 Ae 会逐渐趋近于一个极限值,即总累积排泄量(Ae∞)。利用尿液数据,可以通过 Ae 评估吸收程度,通过最大尿药排泄速率(Rmax)评估吸收速度。例如,氯化钾缓释制剂的BA评价便依赖于测定尿液中钾离子的累积排出量,因为其口服后90%经肾脏排泄,且血液生化系统会自动调节血钾浓度,导致血药浓度测定无法真实反映其吸收过程。
3. 体外评价方法:体内外相关性(IVIVC)与生物豁免
对于缓控释制剂,确立其体外释放度与体内药代动力学参数之间的关联——即体内外相关性(In vitro-in vivo correlation, IVIVC)——是药剂学研究的“圣杯”。一个经过严格验证的IVIVC模型,使得研究者能够利用体外溶出数据预测人体内的BA,从而在处方变更或生产工艺放大时,替代昂贵且耗时的人体临床试验。此外,对于符合生物药剂学分类系统(BCS)I类(高溶、高透)的速释制剂,在满足特定条件时,体外充分的溶出度试验即可直接作为生物等效的依据。
二、实验架构——BA研究的临床试验设计逻辑
获取高质量的药代动力学数据,依赖于严谨、科学的临床试验设计。监管机构(如FDA、NMPA、EMA)对试验设计的每一处细节均有严苛的规范。
1. 核心设计:交叉设计与平行设计
大多数常规制剂的BA/BE研究采用两制剂、两周期、两序列、单次给药的随机交叉设计。这种设计的统计学优势在于:每个受试者在不同的试验周期内分别接受受试制剂和参比制剂,从而使受试者充当自身的对照。这极大地消除了个体间变异对评价结果的干扰。
为防止前一周期药物的残留效应对下一周期产生影响,两次给药之间必须设置足够长的清洗期。药代动力学准则要求,清洗期应不少于目标药物及其活性代谢物消除半衰期的7至10倍。然而,对于半衰期极长(如>24小时)的药物,漫长的清洗期会显著增加受试者的脱落率与依从性风险,此时通常改用单剂量平行设计,即将具有相似人口学特征的受试者分为两组,分别服用不同制剂。
针对体内暴露量波动极大的高变异药物(个体内变异系数CV ≥ 30%),常规的交叉设计难以提供足够的统计效能。此时,必须引入重复交叉设计(Replicate Design),例如三周期部分重复或四周期完全重复设计。这允许研究者准确量化药物的个体内方差,并依法规应用参比制剂标度的平均生物等效性(RSABE)方法进行科学评价。
2. 受试者遴选与剂量设定
受试者群体的选择直接关系到试验结果的外部有效性与内部真实性。一般首选18周岁及以上的健康成年人,要求男女比例均衡,以排除疾病状态对药物吸收代谢的干扰。然而,若受试药物(如抗肿瘤靶向药、强效精神类药物)对健康人群存在不可接受的安全风险,则必须在病情稳定的目标患者群体中进行试验。
在剂量设定上,单次给药BA研究通常采用药品说明书规定的最高规格。这是因为最高规格更能挑战药物在胃肠道的溶解极限。若最高规格存在安全隐患,且该药物在治疗窗口内呈现线性药代动力学特征、各规格处方比例相似且体外溶出曲线一致,方可采用较低规格进行试验。
3. 采样策略的药代动力学考量
血样的采集时间点设计是一门精确的科学,必须完整覆盖药物的吸收相、分布相和消除相。一般单周期需设定12至18个采样点。关键要求包括:
- 在 Tmax 附近设置密集的采样点,以精准捕获真实的 Cmax。
- 末端消除相应至少包含3至4个有效浓度点,以确保采用对数线性回归计算消除速率常数(λz)时的准确度。
- 采样总时长应保证 AUC0-t 能够覆盖外推至无穷大的 AUC0-∞ 的80%以上,确保绝大部分药物吸收过程已被观察。
三、数据解码与影响生物利用度的多维生理因素
在获取原始数据后,研究者需要通过严密的计算公式输出绝对或相对的生物利用度结果,并结合生理学与药剂学原理对数据表现进行解析。
1. 绝对生物利用度与相对生物利用度的数学表达
- 绝对生物利用度(Absolute BA,用 f 表示):用于评估血管外给药(如口服、肌注)与静脉注射(100%直接进入体循环)相比的吸收效率。

- 相对生物利用度(Relative BA,用 F 表示):是评价仿制药质量的核心指标,用于比较受试制剂(T)与已上市原研参比制剂(R)之间的相对吸收程度。
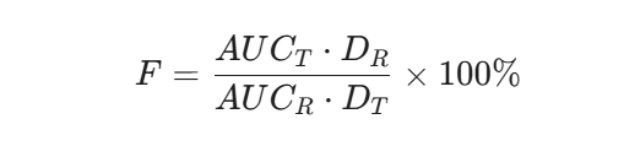
此外,为了剔除吸收程度的干扰、单纯评估吸收速率的差异,学术界建议采用 Cmax/AUC 这一比值指标。该比值仅受吸收速率常数(ka)和消除速率常数(k)的影响,是表征吸收速度的高级动力学参数。
2. 影响药物吸收的内在与外在因素分析
口服固体制剂的BA绝非恒定不变的物理常数,而是一个受制剂特性与人体生理环境双重调控的动态变量。
- 肝脏首过效应:这是限制口服药物绝对生物利用度的最大生理屏障。口服药物在胃肠道被吸收后,必须通过门静脉系统首先进入肝脏。诸如普萘洛尔、利多卡因等药物在首次经过肝脏时,会被肝微粒体细胞色素P450(CYP)酶系大量代谢降解,导致最终进入外周体循环的活性原形药物大幅锐减。
- 胃肠道微环境与代谢:胃肠道内的pH值梯度、消化酶以及肠道菌群,均可能导致药物在被吸收前发生化学或酶促降解。例如,左旋多巴的低BA主要归咎于其在肠黏膜内被芳香L-氨基酸脱羧酶提前代谢。
- 非线性动力学特征:当药物剂量增加导致吸收转运体饱和,或肝脏代谢酶达到饱和(即米氏动力学饱和)时,药物的AUC与剂量将不再呈正比关系。在代谢酶饱和的情况下,大剂量给药可能导致药物避免首过效应,使绝对BA出现不成比例的飙升。
米氏方程
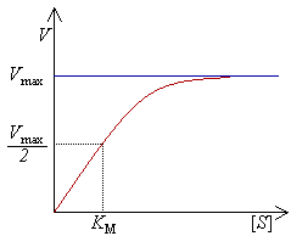
- 食物效应:饮食对药物吸收的干预机制极其复杂。食物可通过延缓胃排空速率(通常降低 Cmax 并延长 Tmax)、刺激胆汁分泌(增加难溶性脂溶性药物的增溶与吸收)、改变肠道内腔pH值、以及增加内脏血流量等方式显著改变BA。为此,法规强制要求采用具有极端挑战性的高热量高脂肪餐(总热量约800-1000 kcal,脂肪供能占50%)来进行进食状态下的BA评价,以暴露制剂可能存在的最大“食物-药物相互作用”缺陷。
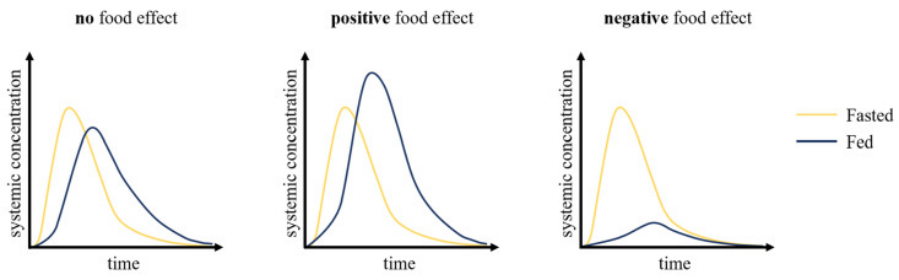
药物在空腹和进食状态下的药代动力学特征。在无食物影响的情况下,药代动力学特征的变化很小,且在口服给药后观察到的正常波动范围内。食物的积极作用与全身药物暴露量的增加相关,而食物的消极作用与药物暴露量的降低相关。(doi: 10.3390/pharmaceutics12070672)
四、制剂复杂性——特殊剂型的BA评价策略
随着制药工程技术的迭代,越来越多的药物被开发成复杂的递送系统。传统的药代动力学评价方法难以全面刻画这些特殊制剂的体内行为,必须为其量身定制评价方案。
1. 口服缓释与控释制剂
缓控释制剂通过高分子骨架、渗透泵或微丸包衣等先进技术,人为改变了活性成分的释药动力学,旨在减少给药频次、平稳血药浓度。其BA评价不仅要求单次给药试验,更强制要求进行多次给药(稳态)试验。
- 单次给药空腹/餐后试验:核心目的是验证其缓释特征的真实性。通过与普通速释制剂对比,合格的缓控释制剂应表现出相对BA(基于AUC)不低于普通制剂的80%,但其 Cmax 必须被显著压低,且 Tmax 显著延迟。
- 多次给药稳态评价:受试者需按临床说明书推荐的给药间隔,连续服药至少7个半衰期。通过在末次给药前连续三天测定谷浓度(Cmin)不变,以确认体内已达到动力学稳态。稳态下的关键评价参数不仅包括稳态药时曲线下面积(AUCτ)、平均稳态血药浓度(Cav = AUCτ/ τ),更引入了血药浓度波动度(Degree of Fluctuation, DF)这一核心指标。计算公式为:
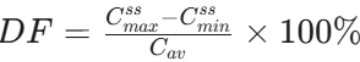
优秀的缓控释制剂应在保证生物利用度的同时,展现出显著优于普通制剂的(即更低的)波动度,从而验证其削峰填谷的临床设计初衷。
2. 复方制剂(FDCs)
现代临床治疗日益倾向于使用包含两种或多种活性成分的复方制剂以提高患者依从性。复方制剂的BA评价,其本质是进行一次人体内部的药物-药物相互作用(DDI)及处方合理性考核。
常规的设计方案是:将最高规格的复方制剂作为受试药物,将分别等剂量的各单方制剂联合服用作为参比方案,进行随机交叉对照试验。研究者必须独立计算复方中每一种活性成分的药代动力学参数(AUC、Cmax等),并分别论证其等效性。如果在特定情况下,复方中的某一组分(辅药)仅仅是为了通过抑制代谢酶来提升主药的生物利用度(例如:某些蛋白酶抑制剂复合制剂中使用利托那韦作为药代动力学增强剂),且辅药本身无直接治疗目的或已是成熟药物,评价的重心则可向主药的暴露量变化倾斜。
五、结语
生物利用度(BA)研究不仅是横亘在新药研发与仿制药一致性评价中的一道科学门槛,更是连接实验室药学指标与真实世界临床疗效的关键纽带。从受试者的入组排查,到高精度液相色谱-质谱联用(LC-MS/MS)分析,再到复杂的非房室模型数学推演,BA评价的每一个环节都凝结着极高的技术密度与法规要求。深入理解生物利用度的评价体系及其背后的生理药剂学机制,对于推动制药工艺革新、优化个体化给药方案、最终保障公众的用药安全与有效,具有无可替代的深远意义。
扩展阅读:
1. 解锁口服药物吸收密码:溶解性、渗透性及S-P矛盾全解析
2. 抗抑郁药沃替西汀研发全解:多模式机制、晶型奥秘与早期原料药开发大揭秘
3. 创新药降本增效的核心密码:深度解析现代工艺开发新技术,合成生物化学等三大工艺成核心驱动力!
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论