

































在癌症治疗的漫长征途中,肺癌,尤其是非小细胞肺癌(NSCLC),一直是一座难以逾越的大山。然而,随着对癌症分子机制理解的深入,靶向治疗为患者带来了希望的曙光。本章将带您深入了解一种具有里程碑意义的药物——色瑞替尼(Ceritinib, 研发代号LDK378, 商品名Zykadia®),它是如何从实验室的奇思妙想,演变为战胜克唑替尼耐药性的强力武器。
一、初识ALK与癌症的纠葛
间变性淋巴瘤激酶(ALK)本是人体内一种正常的受体酪氨酸激酶,在正常组织中,其表达主要局限于神经系统的特定亚群,且在哺乳动物发育过程中的确切作用尚不完全清楚。科学界甚至通过敲除小鼠的ALK基因,发现其能正常发育,仅表现出一些抗抑郁特征和海马体相关能力的增强。
然而,当ALK遇到遗传异常,情况便急转直下。这些异常包括激酶结构域与多种伴侣基因发生易位(融合),或导致非配体依赖性组成性激活的激活突变。
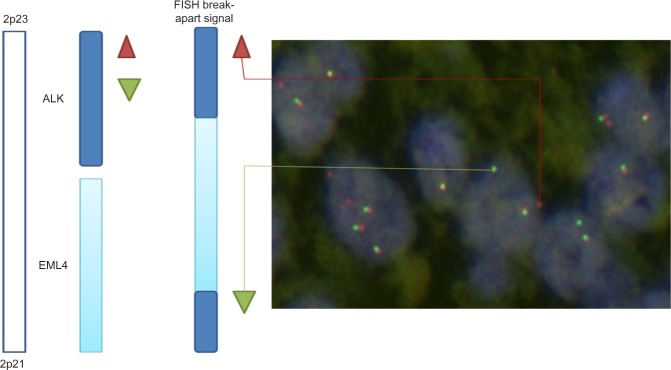
ALK的这种失调最初是在间变性大细胞淋巴瘤(ALCL)中被发现的,表现为NPM-ALK融合蛋白。随后,科学家们在包括炎性肌纤维母细胞肿瘤(IMT)、弥漫性大B细胞淋巴瘤(DLBCL)以及至关重要的、约2%~7%的非小细胞肺癌(NSCLC)患者中,都发现了导致ALK与各种伴侣基因融合的染色体重排。在NSCLC中,EML4是ALK最主要的融合伴侣。
非小细胞肺癌中EML4-ALK融合癌基因的示意图及FISH检测
二、破局耐药,色瑞替尼的设计智慧
克唑替尼(Crizotinib)作为第一代ALK抑制剂,曾给ALK阳性NSCLC患者带来了巨大的临床获益。然而,好景不长,患者通常在治疗10~11个月后不可避免地出现耐药突变,导致药物失效。面对这一临床痛点,研发更为有效、能够克服耐药的新一代ALK抑制剂迫在眉睫。
代谢之殇:TAE684的遗憾
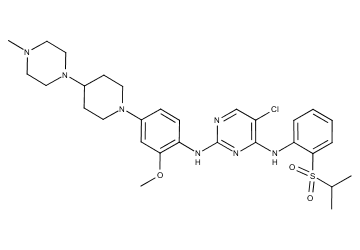
早期的研发目光聚焦在如TAE684(2006年公开的第一个有效的ALK抑制剂)及其类似物GNF0912等强效ALK抑制剂上。尽管它们在体外表现优异,但临床前评估揭示了一个严重的毒性隐患:这些化合物在体内通过代谢氧化,会形成大量的反应性加合物。
TAE684化学结构
半定量分析显示,约20%的TAE684和GNF0912会被转化为反应性物质。这些活性物质虽然可以通过谷胱甘肽捕获实验被监测到,但推测其可能与肝毒性及其他特定毒性有关。在TAE684的毒理学评估中,更是发现了不可逆毒性。因此,消除这些反应性代谢物的形成,成为了药物化学家们的首要任务。
构效关系的启示
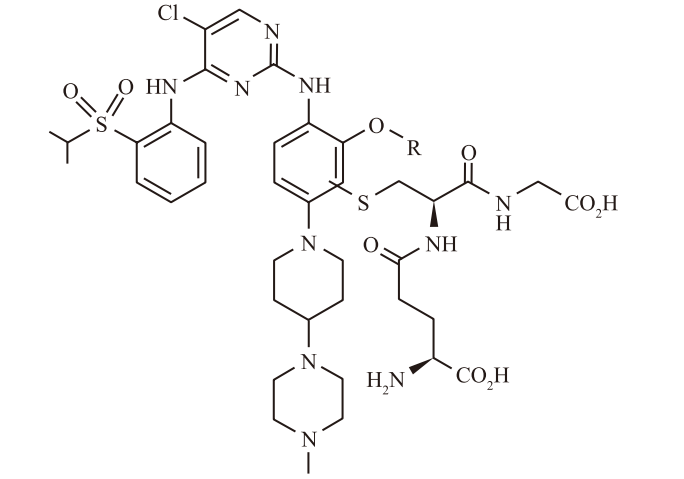
通过系统的构效关系(SAR)评估,研究人员揭示了一个关键信息:反应性代谢物的形成,主要与通过氮原子连接到苯胺上的水溶性基团的存在有关。推测富电子的芳环通过代谢氧化形成了高度反应性的1,4-二亚氨基醌,该结构在谷胱甘肽(GSH)存在下会形成GSH加合物,从而产生毒性。
GSH加合物化学结构
为了消除这种潜在毒性,研究人员提出了精妙的设计策略。他们决定将TAE684和GNF0912中的哌啶环反转,从而切断形成推测的二亚氨基醌的途径。此外,他们还在烷氧基的对位引入了甲基,以阻止该位置的进一步代谢。
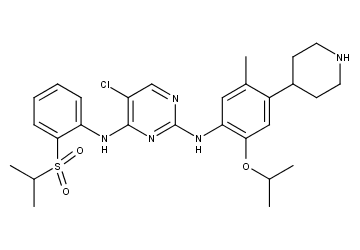
这些基于代谢机制理解的结构优化,最终导向了活性最优、且显著降低了反应性代谢物形成风险的色瑞替尼。
色瑞替尼化学结构
这一精妙的设计策略,不仅保留了对ALK的强效抑制活性,更解决了困扰早期候选药物的代谢毒性问题,为色瑞替尼走入临床奠定了坚实基础。
三、色瑞替尼的诞生:高效的汇聚性合成
在确立了分子的终极形态后,设计一条高效、高产率且具备大规模工业化生产潜力的合成路线成为了当务之急。色瑞替尼的合成彻底摒弃了冗长低效的直线型合成,转而采用了一种极其优雅的“高度汇聚性”策略。该路线以结构对称且反应活性层次分明的2,4,5-三氯嘧啶作为核心连接枢纽,通过控制反应温度和催化条件,实现两个连续胺化反应。
整个合成网络被清晰地划分为两大独立并行的支线任务: 第一条支线负责构建左侧的磺酰基苯胺片段。以氟硝基苯为初始原料,在碳酸钾催化下与丙烷-2-硫醇发生亲核取代,随后利用过硼酸钠的强氧化性将硫醚转化为砜,最后通过催化加氢还原硝基,以极高的总收率获得关键中间体1:2-(异丙基磺酰基)苯胺。
第二条支线则负责构建右侧复杂的哌啶基苯胺片段。以2-氯-4-氟甲苯为起点,经过一系列精密的官能团转化,包括异丙氧基的引入、硝化反应,以及最关键的BOC保护的哌啶环的Suzuki偶联或类似碳碳键构建反应,最终得到核心中间体2:2-异丙氧基-5-甲基-4-(哌啶-4-基)苯胺。
在最终的组装阶段,中间体1首先在较低温度下与2,4,5-三氯嘧啶的C4位氯原子发生特异性取代;随后,在酸性条件及微波加热(或高温回流)的促发下,引入中间体2替换C2位的氯原子。最后一步利用无水氯化氢的二氧六环溶液脱除哌啶环上的BOC保护基,即可获得高纯度的色瑞替尼盐酸盐。这种汇聚式路线不仅显著缩短了最长线性步骤的长度,极大地提高了原药的总收率,且有效控制了杂质的生成,为色瑞替尼的快速临床前储备和后期的商业化量产奠定了坚不可摧的物质基础。
四、深度的临床前图谱——药效、选择性与药代动力学特征
从实验室的烧瓶中诞生后,色瑞替尼面临着一系列极其严苛的临床前测试。数据证明,这款基于理性设计原则雕琢而成的分子,在药理学特性的每一个维度上都交出了近乎完美的答卷。
极具威慑力的ALK抑制活性
体外活性评估有力地证实了色瑞替尼是一种极其有效的ALK抑制剂。
在针对纯化的ALK蛋白、表达NPM-ALK或EML4-ALK融合蛋白的Ba/F3细胞,以及经ALK重排的肺癌细胞株H2228和H3122的测试中,色瑞替尼都表现出了极低的IC50值(0.15 nmol/L至22.0 nmol/L不等)。此外,它还能有效抑制karpas299细胞中ALK的磷酸化及其下游信号通路,从源头上切断癌细胞的生长信号。
ALK-crizotinib耐药突变的 Ba/F3 模型
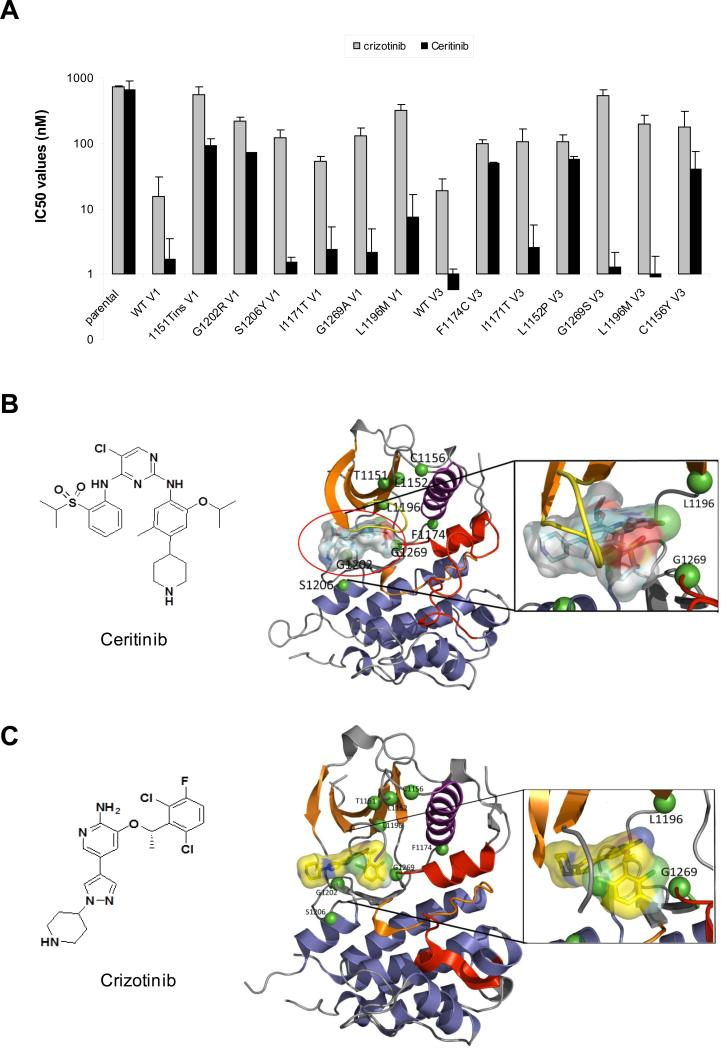
卓越的激酶选择性:精准打击,避免误伤
一个优秀的靶向药物不仅要强效,更要精准。在对46种激酶的筛选测试中,色瑞替尼展现出了优异的选择性。
在所测试的众多激酶中,色瑞替尼仅对三种激酶(IGF-1R、InsR和STK22D)表现出低于100 nmol/L的抑制活性(IC50分别为8 nmol/L、7 nmol/L和23 nmol/L)。然而,由于色瑞替尼对主靶点ALK的抑制活性达到了惊人的200 pmol/L,因此对这些脱靶蛋白的选择性分别为80倍、70 倍和230倍。
在进一步的细胞水平选择性测试中,除了ALK,在所有测试激酶中均未观察到IC50低于100 nmol/L的抑制作用,再次验证了其卓越的选择性,降低了因广泛脱靶而引起毒副作用的风险。
临床前药代动力学:持久与稳定
色瑞替尼在小鼠、大鼠、犬和猴等多种动物模型中,展现出了令人满意的药代动力学(PK)特征:
- 清除率:在各种物种中均较低。
- 分布容积:稳态分配容积(Vss)较高,约为体液总量的10倍,表明药物能广泛分布于组织中。
- 半衰期:从中等到较长(小鼠6.2 h,猴26 h),有利于药物在体内维持有效浓度。
- 生物利用度:无论口服溶液还是混悬液,在所有物种中均表现出良好的口服生物利用度(>54%)。
这些优良的PK性质,预示着色瑞替尼在体内具有持久、稳定的抗肿瘤作用,为临床给药方案的制定提供了重要依据。
体内药效:让肿瘤消退的强力一击
在小鼠异种移植模型中,色瑞替尼的体内药效评价取得了振奋人心的结果。重点聚焦于两种模型:Karpas299(具有NPM-ALK融合)和H2228(具有EML4-ALK融合)。
色瑞替尼在小鼠H2228和Karpas299异种移植模型中的活性
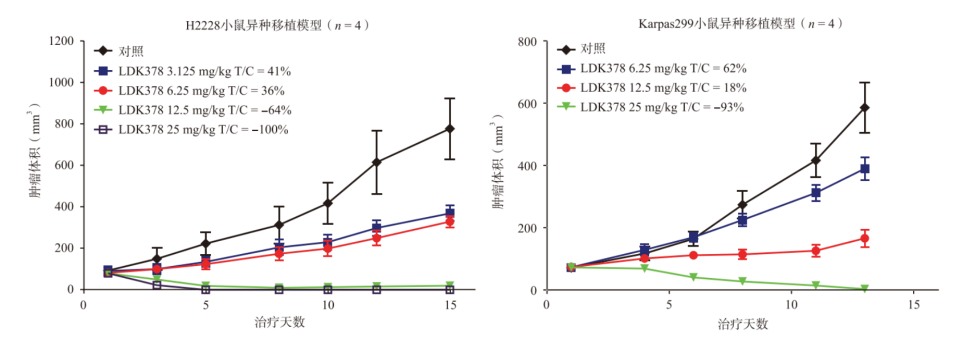
在为期2周的实验中,色瑞替尼表现出了令人印象深刻的抗肿瘤活性:
- 剂量依赖性肿瘤生长抑制:在H2228模型中,低剂量即可抑制肿瘤生长,而较高剂量(12.5 mg/kg和25 mg/kg)则诱导了显著的肿瘤消退,消退率分别达-64%和-100%。在Karpas299模型中,同样观察到剂量依赖性的抑制,25 mg/kg剂量下肿瘤几乎完全消退(-93%)。
- 耐受性良好:在所有测试剂量下,均未观察到小鼠体重减轻,表明药物在强效抗肿瘤的同时,毒副作用可控。
- 药效持久:为了评估药效持续时间,研究人员在H2228小鼠模型中以50 mg/kg剂量给药2周后停止,结果显示色瑞替尼表现出持久的抗肿瘤活性,药效超过150天,大大延长了肿瘤复发的时间。
五、跨越耐药的鸿沟——破解克唑替尼的耐药密码
色瑞替尼作为第二代ALK抑制剂,其最核心的临床价值在于能够克服第一代药物克唑替尼所产生的耐药性。在克唑替尼获得加速批准的同时,研发小组未雨绸缪,决定评估色瑞替尼对克唑替尼耐药突变的活性,旨在为这部分患者提供新的治疗选择。
为了模拟临床耐药情况,研究人员采用了具有两个最常见EML4-ALK突变(L1196M和G1269A)的克唑替尼耐药细胞系模型。其中,H312 CR1是一种由于长期暴露于克唑替尼而产生的耐药细胞系,具有L1196M看门基因突变和EML4-ALK等位基因扩增。
此外,还使用了来自对克唑替尼耐药的ALK重排肺癌患者活检组织建立的细胞系。实验有力地证实,色瑞替尼在这些主要的抗性突变(L1196M、G1269A、S1206Y、I1171T)细胞中均显示出良好的药效。
通过深入分析色瑞替尼与ALK激酶域的共结晶结构,我们可以清楚地理解克唑替尼产生耐药、而色瑞替尼为何能克服这些耐药突变的分子机制。
这一结构层面的清晰阐释,不仅增强了我们对克唑替尼耐药机制的理解,更从分子层面确立了色瑞替尼作为第二代ALK抑制剂无可替代的地位。
研究人员在多种克唑替尼耐药的异种移植肿瘤模型中,对色瑞替尼的活性进行了测试。
结果令人振奋:色瑞替尼在C1156Y和I1171T耐药模型中均展现出强效的抗肿瘤活性。通过其他模型的进一步验证,表明色瑞替尼在所有测试的耐药肿瘤模型中均表现出一定的活性。只有G1202R突变对色瑞替尼最不敏感(数据未显示),这也印证了前述晶体结构分析中关于空间冲突的推测。
六、“36个月”的医药界传奇——ASCEND系列临床试验与生存奇迹
基于无可挑剔的临床前抗耐药数据,诺华公司迅速启动了色瑞替尼的全球临床开发计划。
震撼业界的ASCEND-1 I期临床数据
新药的I期临床试验通常仅以探索安全性和最大耐受剂量(MTD)为首要目标,但在ASCEND-1试验中,色瑞替尼不仅确立了每日口服750 mg的MTD,更在剂量扩展阶段爆发出令整个肿瘤学界震撼的治疗效力。
在Ⅰ期临床研究期间,共治疗了130名患者,其中114例剂量≥400 mg/d。在先前接受克唑替尼治疗的部分患者中(66例),ORR为 57%;在未曾接受过克唑替尼治疗的患者中(35例),ORR为60%;所有患者的 ORR为58%。
ASCEND-1 I期临床研究中色瑞替尼的临床缓解率总结(400 ~ 750 mg/d)
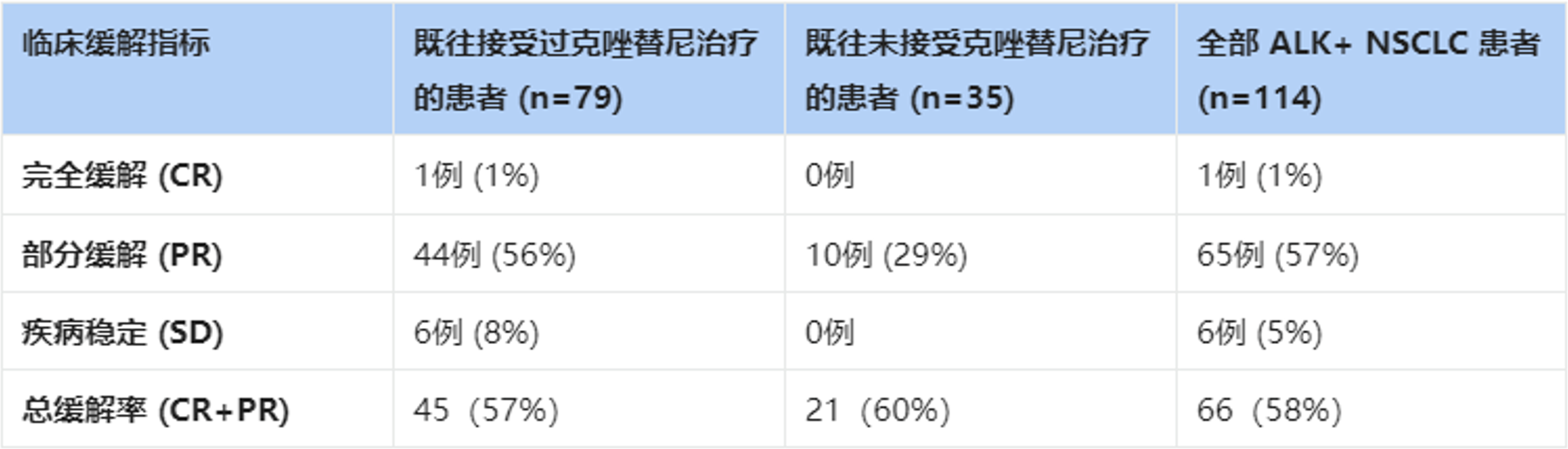
(数据来源:ASCEND-1临床研究初步结果展示,无论是否有过耐药史,患者均能获得极高比例的深度缓解。)
对于晚期ALK阳性肺癌患者而言,中枢神经系统(特别是脑实质)是肿瘤细胞极易发生转移的“避风港”。由于血脑屏障的阻挡,第一代药物克唑替尼在脑部的药物浓度极低,导致大量患者最终死于脑转移。ASCEND-1研究的深入分析表明,色瑞替尼不仅能够强效缩减全身脏器的病灶,更展现出了优异的颅内穿透能力和抗肿瘤活性,为伴有脑转移或脑膜转移的危重患者提供了实质性的生存获益。在安全性评估中,色瑞替尼最常见的不良反应主要集中在胃肠道系统(如腹泻、恶心、呕吐)及肝转氨酶的无症状升高。这些不良事件绝大多数属于1至2级,具有高度可逆性,且通过短暂停药或降低剂量(如降至600 mg或450 mg并随餐服用)即可得到有效管理,因毒性导致永久停药的比例极低。
极速的“突破性疗法”审批与全面碾压化疗的ASCEND-4/5
面对这种对患者生存期有着根本性挽救作用的颠覆性疗效,美国FDA展现出了罕见的政策灵活性与极高的审评效率。2013年3月,FDA基于I期临床的惊艳数据,破例授予色瑞替尼“突破性疗法”认定。2014年4月,FDA正式启动加速审批通道,批准色瑞替尼上市,专门用于治疗克唑替尼耐药或不耐受的ALK阳性转移性NSCLC患者。
从2011年1月首例患者入组I期临床试验,到2014年4月成功获批商业化上市,色瑞替尼仅仅耗时36个月。在平均耗时10年以上且充满死亡风险的新药研发领域,这不仅是一项难以企及的竞速奇迹,更是精准医疗、靶向设计与监管创新完美结合的最高赞歌。
上市获批仅仅是攀登的起点。在随后开展的极其严谨的全球多中心、随机、对照III期临床试验(ASCEND-4和ASCEND-5)中,色瑞替尼更是以绝对的实力碾压了传统的标准化学治疗方案。 在ASCEND-4研究中(针对初治的ALK阳性晚期患者),接受色瑞替尼一线治疗的患者由独立盲法评审委员会(BIRC)评估的中位无进展生存期(PFS)达到了惊人的16.6个月,而对照组的标准含铂双药化疗联合培美曲塞维持治疗仅为8.1个月。色瑞替尼不仅使疾病进展或死亡的风险暴降了45% (HR=0.55, p<0.001),更将客观缓解率从化疗组的27%大幅提升至73%。
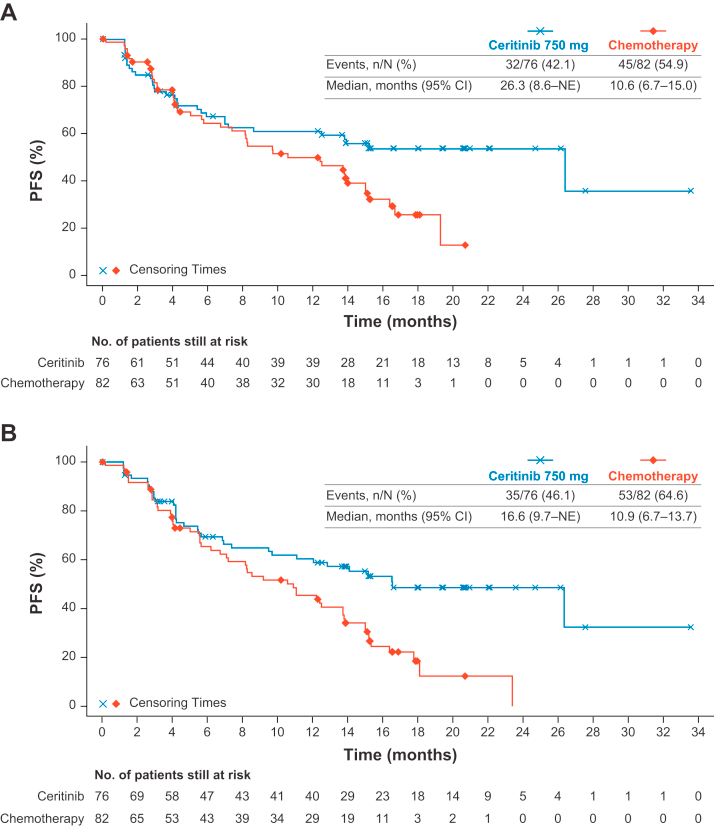
Kaplan-Meier曲线图显示了根据(A) BIRC评估和(B)研究者评估得出的PFS。BIRC,盲法独立审查委员会;CI,置信区间;n,纳入分析的事件总数;N,纳入分析的患者总数;NE,无法估计;PFS,无进展生存期。
在ASCEND-5研究中(针对已经接受过化疗及克唑替尼双重治疗且失败的重度耐药患者),色瑞替尼依然展现出强大的底蕴。其PFS显著优于化疗(5.4个月 vs 1.6个月,HR=0.49, p<0.001),且客观缓解率实现了对化疗的降维打击(39.1% vs 6.9%)。这些确凿的III期临床铁证,使得FDA在2017年进一步拓展了色瑞替尼的适应症,将其正式推上了ALK阳性肺癌一线治疗的王者宝座。
七、群星璀璨的竞争格局与排兵布阵
随着医学科技的狂飙突进,ALK阳性肺癌的治疗领域迎来了真正的群星璀璨时代。 通过摩熵医药数据库的竞争情报系统透视这一赛道,在色瑞替尼成功突围之后,以阿来替尼(Alectinib)、布加替尼(Brigatinib)为代表的其他第二代ALK抑制剂,以及专门针对终极耐药突变G1202R设计的第三代抑制剂劳拉替尼(Lorlatinib)相继涌现,彻底重塑了临床治疗的竞争格局。
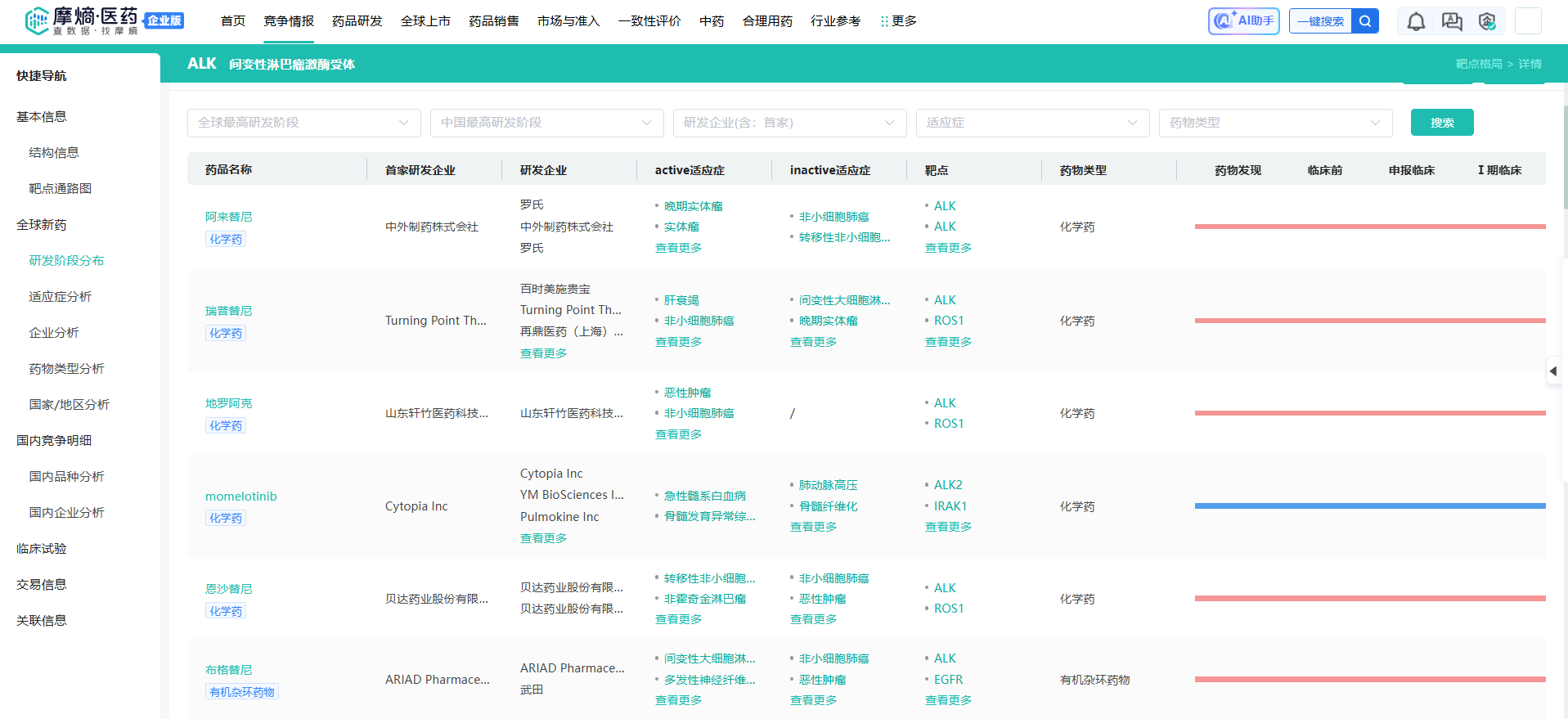
查数据,找摩熵!图源:摩熵医药全球药物研发数据库
在这一激烈的竞争版图中,各项大型临床试验(如ALEX、J-ALEX等)不断刷新着患者的生存上限。例如,阿来替尼在一线治疗的头对头比较研究(ALEX试验)中展现出了极其优异的疗效,其PFS突破了25个月,且由于其独特的高脂溶性,阿来替尼在穿透血脑屏障控制脑转移方面表现出类拔萃。更重要的是,与色瑞替尼在750 mg空腹给药方案下较高的胃肠道不良反应发生率相比,阿来替尼表现出了更好的耐受性,使其迅速确立了新的一线标准治疗地位。
然而,这并不意味着色瑞替尼退出了历史舞台。相反,真实世界的数据与临床经验表明,色瑞替尼在精准排兵布阵中依然扮演着不可替代的战略角色。许多临床医生(如马萨诸塞州综合医院的Alice Shaw博士)在长期实践中发现,通过优化给药方案(如降低剂量至450 mg并随餐饮用),可以在毫不妥协疗效的前提下,极大地减轻色瑞替尼的胃肠道毒性,显著提升患者的生活质量。此外,当患者在使用其他二代药物(如阿来替尼)进展时,若肿瘤细胞演化出特定的激酶构象变化,色瑞替尼仍可作为重要的后线拯救治疗手段。这场不同代际、不同分子结构抑制剂之间的接力赛,最终的获益者是那些曾经绝望的癌症患者。
八、结语
色瑞替尼的诞生,是基于对早期候选药物代谢机制的深入理解、精妙的药物分子设计以及严谨的临床前和临床评估的结晶。它从Ⅰ期临床试验到获批上市,仅耗时短短36个月,这一惊人的速度凸显了其巨大的临床价值和未满足的医疗需求。
作为首个被FDA认定为突破性疗法并获批的第二代ALK抑制剂,色瑞替尼的出现,不仅为对克唑替尼停止响应的ALK阳性NSCLC患者提供了新的、更为有效的替代疗法,更深刻地改变了这部分患者的预后。它的研发历程,是一篇基于代谢机制理解进行逆袭、从克服毒性到攻克耐药的成功典范,必将载入癌症治疗的史册。
扩展阅读:
1. 前列腺癌非甾体AR拮抗剂进化史:氟他胺破冰、比卡鲁胺称王、恩扎卢胺重塑生存期
2. 匹托利生:从硫丙咪胺的肝毒性到全球首款H3受体反向激动剂,攻克血脑屏障,改写嗜睡症治疗指南
3. 创新药降本增效的核心密码:深度解析现代工艺开发新技术,合成生物化学等三大工艺成核心驱动力!
查数据,找摩熵!想要解锁更多药品信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!













 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论