

































在抗肿瘤药物的研发史上,有一类故事最迷人:死灰复燃。
不是所有的新药都诞生于全新的靶点。有时候,宝藏就埋在几十年前被遗忘的“废纸堆”里。今天我要讲述的主角——曲氟尿苷-替匹嘧啶(FTD-TPI,LONSURF),就是这样一个教科书级别的案例。它讲述了科学家如何通过“伴侣药物”的策略,拯救了一个半衰期只有18分钟的“失败”分子,并最终将其推向全球市场的传奇经历。
一、Heidelberger的设想:5-FU与FTD
故事要从化疗界的“基石”——5-氟尿嘧啶(5-FU)说起。自从1957年Heidelberger博士利用肿瘤细胞代谢上调的特性合成5-FU以来,它就一直是结直肠癌治疗的顶梁柱 。设计理念简单而优雅:利用癌细胞快速增殖对核酸合成的巨大需求,引入氟原子伪装成尿嘧啶,诱骗癌细胞摄取。
但是,5-FU有个致命弱点:它极易被体内的二氢嘧啶脱氢酶(DPD)降解。为了解决这个问题,药学家们不仅开发了静脉注射剂,还搞出了一系列口服前药(如卡培他滨、替加氟),甚至给它找了“保镖”(如UFT和TS-1中的DPD抑制剂),目的只有一个:抑制降解,提高浓度 。
就在这个时期,Heidelberger博士其实还留了一手。1964年,他合成了另一个核苷类似物——曲氟尿苷(FTD) 。
FTD在体外实验中表现极其强悍,甚至对那些已经对5-FU耐药的癌细胞都有效 。按理说,这应该是一个超级重磅炸弹。但在临床试验中,FTD遭遇了滑铁卢。
为什么?因为它太“短命”了。
FTD在人体内的血浆半衰期只有短短的18分钟 。就像一阵风,还没来得及杀伤肿瘤,就被肝脏首过效应和肠道中的胸苷磷酸化酶(TP)切碎代谢掉了。为了维持有效浓度,医生不得不让患者每天连续输注,副作用大到无法承受。最终,FTD被迫退出了静脉给药的舞台,只能委身于眼科,作为一种抗病毒滴眼液(商品名Viroptic)用于治疗单纯疱疹病毒性角膜炎,因为局部给药可以避开肝脏的首过效应 。
二、替匹嘧啶(TPI)的诞生
1. 研发理念的复活
时间来到20世纪90年代。大鹏制药的研究团队在成功开发了UFT和S-1后,积累了丰富的“酶抑制剂复方”开发经验。他们重新审视了FTD这把被遗弃的利剑。
逻辑很简单:既然S-1通过抑制DPD成功复活了5-FU,那么我们是否可以通过抑制TP酶来复活FTD?
如果能找到一种强效、特异性的TP抑制剂,就能阻断FTD的快速降解,将其半衰期从18分钟延长到具有临床意义的水平,从而释放其强大的抗肿瘤潜力。
2. 跨越物种的陷阱
寻找TP抑制剂的过程并非一帆风顺,早期研究遭遇了严重的“种属差异”陷阱。
在生物学研究中,常用的小鼠、大鼠等啮齿动物,其体内的酶谱与人类截然不同。
- 人类:TP活性极高,尿苷磷酸化酶(UP)活性较低。FTD主要被TP降解。
- 啮齿动物:UP活性极高,TP活性较低。
早期的研究者在筛选抑制剂时,往往使用大肠杆菌或啮齿动物的酶作为模型。结果发现,筛选出的化合物(如BAU等非环状尿苷类)在动物身上有效,但在人身上却失效了——因为它们主要抑制的是UP,而非TP,或者对人源TP的结合力太弱 。
痛定思痛,大鹏制药的研究团队做出了一个关键决定:必须直接使用纯化的人源TP蛋白进行抑制剂的筛选。
3. 分子雕刻:从6A5CU到TPI
研究团队以 6-氨基-5-氯尿嘧啶(6A5CU)为先导化合物,开启了漫长的结构修饰之路。6A5CU对人TP有一定的抑制作用,但活性不够强(IC50为1.5 ×10-5mol/L),且缺乏选择性,同时抑制UP 。
为了获得完美的抑制剂,化学家们对嘧啶环进行了精细的“手术”,重点考察了C-5位和C-6位的取代基效应。
表2.1:关键筛选化合物对人TP及大鼠UP的抑制活性对比
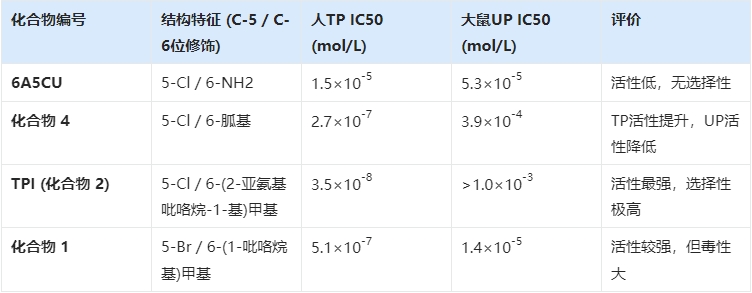
筛选过程的深度解析:
(1)C-6位的突破:研究人员发现,在C-6位引入含有氮原子的杂环结构能显著提高与TP酶活性中心的结合力。特别是引入(2-亚氨基-1-吡咯烷-1-基)甲基这一结构时,抑制活性发生了质的飞跃,IC50值达到了纳摩尔级别(3.5×10-8mol/L)。更重要的是,这一结构对UP酶几乎没有抑制作用(IC50 > 10-3mol/L),实现了极高的选择性。这意味着它只保护FTD,而不干扰其他尿苷类物质的代谢。
(2)C-5位的微调:在确定了C-6位的最佳基团后,研究人员考察了C-5位卤素的影响。实验表明,氯(Cl)、溴(Br)或碘(I)都能保持高活性,但如果是甲基,活性则大幅下降。
4. 安全性的抉择:放弃“最强”,选择“最稳”
在最后的决赛圈中,化合物1(5-溴衍生物)和化合物2(即TPI,5-氯衍生物)展开了角逐。
虽然化合物1在小鼠体内的药代动力学表现略优(AUC更高),但在临床前安全性评价中,研究人员发现了一个令人担忧的信号:化合物1显示出明显的神经毒性。这可能是由于其脱靶效应导致的。
相比之下,TPI(化合物2)虽然活性略低,但安全性极佳,没有观察到神经毒性。在药物研发中,安全性往往是一票否决权。最终,大鹏制药选择了TPI作为FTD的“守护者”。
三、黄金比例 1 : 0.5
有了“矛”(FTD)和“盾”(TPI),接下来的问题是:怎么配比?
这可不是简单的1+1。研究人员在猴子体内进行了详细的药代动力学实验。研究人员测试了FTD与不同浓度TPI的组合(摩尔比 1:0.1, 1:0.2, 1:0.5, 1:1, 1:2 等)。
研究表明,在 1 : 0.5 的比例下,体内的TP酶已经被有效地饱和抑制,再增加TPI不仅浪费,还可能增加身体负担。
随后,研究人员在人胃肠道癌细胞(CO-3)异种移植的裸鼠模型中验证了这一比例的抗肿瘤效果。
表 3.1:不同配比对小鼠肿瘤生长的抑制作用
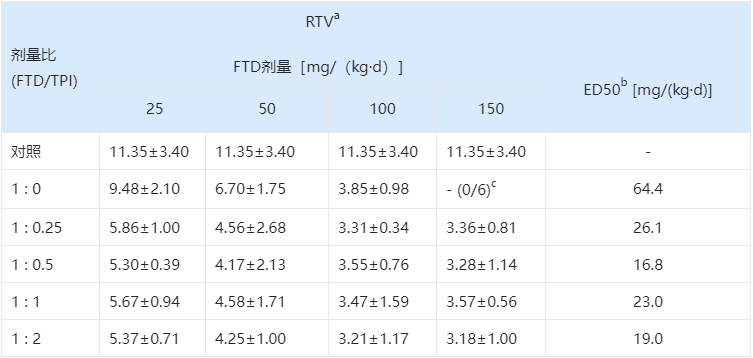
a相对肿瘤体积=(第15天的肿瘤体积)/(第0天的肿瘤体积)。
b引起50%的肿瘤生长抑制所需的FTD剂量。
c FTD单用治疗组中的6只受试小鼠,在100 mg/kg和150 mg/kg的每日剂量下,分别有1只和6只因FTD 的毒性而过早死亡。
实验结果显示,单用高剂量FTD虽然能抑制肿瘤,但会导致小鼠因毒性死亡。而1:0.5的组合不仅实现了最佳的肿瘤抑制(RTV最小),且小鼠耐受性良好。
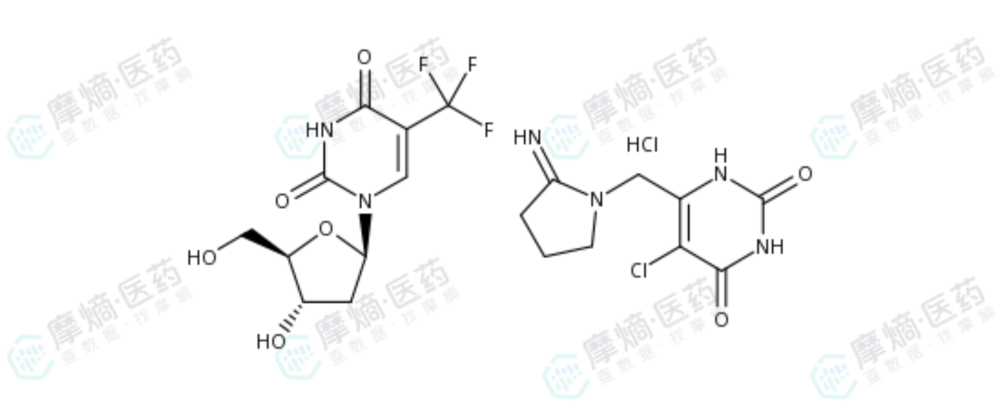
最终,1 : 0.5(摩尔比)被确定为TAS-102的固定配方。这一黄金比例在后续的所有临床试验及商业化产品中被严格沿用。
TAS-102结构式
四、独特的机制
很多人会误以为FTD-TPI只是另一个5-FU。这是巨大的误解。
虽然它们都是氟嘧啶类药物,但FTD-TPI的杀手锏完全不同:5-FU:主要是通过抑制TS酶,阻断DNA合成原料,同时整合进RNA造成功能紊乱。
FTD-TPI:它更像是一个“DNA伪装者”。
- FTD在细胞内被磷酸化后,生成的F3dTTP会直接整合进癌细胞的DNA链中。
- 一旦嵌入,DNA的功能就会受损,甚至导致双链断裂。
- 更绝的是,这种嵌入是非常“顽固”的。研究发现,负责修复DNA的糖基化酶(如UNG, SMUG1)根本切不动整合了FTD的DNA链。
TAS - 102与5 - FU作用机制的比较
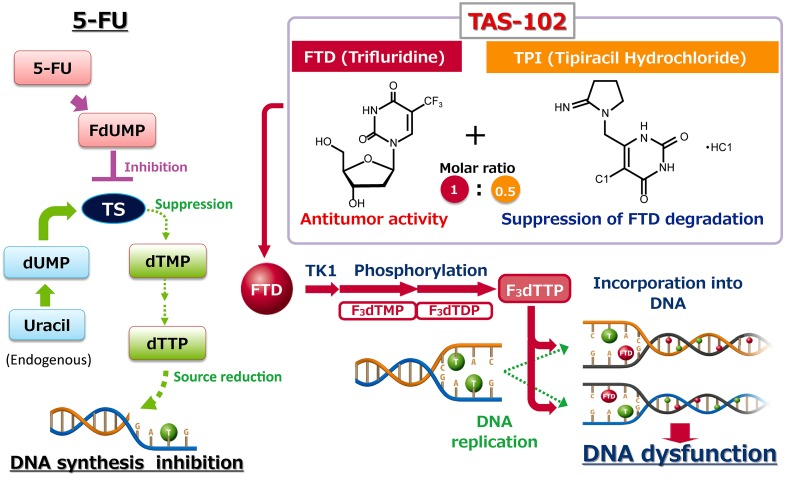
结论:FTD-TPI的核心机制是DNA损伤,而非单纯的酶抑制。这也是为什么它能对5-FU耐药的肿瘤依然有效的原因——因为它换了一条赛道攻击癌细胞 。
五、从实验室到临床的跨越
FTD-TPI的临床之路并非一帆风顺。
初战失利:在美国最早的I期临床中,采用每日给药方案,结果完全没看到肿瘤缓解,且疾病稳定期很短。
调整战术:研究人员回到动物模型,发现分次给药(每天2次)比每天1次更能增加FTD在DNA中的整合量。
最终方案:他们设计了独特的“5天吃药,2天休息”(2周一个循环,然后休整2周)的方案。这个节奏既保证了药物持续嵌入DNA,又给了骨髓喘息的时间。
采用新方案后,日本的I期临床试验取得了突破。确定了推荐剂量(RP2D)为 35mg/m2/次,每日两次。
随后的J003 II期临床研究更是给出了令人振奋的数据:在169名标准治疗失败的难治性转移性结直肠癌患者中,TAS-102组的中位总生存期(OS)达到9.0个月,而安慰剂组仅为6.6个月。死亡风险降低了44%。
这一结果不仅让TAS-102在日本率先获批,更为其走向全球奠定了坚实基础。
六、RECOURSE与TERRA的双重奏
为了获得美国FDA和欧洲EMA的认可,TAS-102 必须在全球范围内证明自己。大鹏制药启动了两项规模宏大的III期临床试验:针对全球人群的RECOURSE和针对亚洲人群的TERRA。
1. RECOURSE研究:确立全球标准
研究背景:这是一项全球多中心、随机、双盲、安慰剂对照的III期研究,覆盖了北美、欧洲、日本和澳大利亚。
入组患者:800名“绝境”患者。这些患者此前已经接受了所有可用的标准治疗(包括氟嘧啶、奥沙利铂、伊立替康、贝伐珠单抗,以及抗EGFR抗体),但病情依然进展。在TAS-102之前,他们已无药可救。
关键数据:
总生存期(OS):TAS-102组 7.1个月 vs 安慰剂组 5.3个月。
风险比(HR):0.68。这意味着TAS-102将死亡风险降低了32%(P < 0.001)。
亚组一致性:无论患者之前的治疗史如何(是否用过靶向药),无论KRAS基因是野生型还是突变型,TAS-102都显示出了生存获益。
这一“硬核”数据,直接叩开了FDA的大门。2015年9月,美国FDA批准TAS-102上市,用于治疗难治性转移性结直肠癌。
2. TERRA研究:亚洲人群的验证
考虑到东西方人群在药物代谢和耐受性上可能存在差异,大鹏制药在中国、韩国和泰国开展了TERRA研究 。
关键数据:
总生存期(OS):TAS-102组 7.8个月 vs 安慰剂组 7.1个月。
风险比(HR):0.79(P = 0.0035)。
TERRA研究的成功,不仅验证了TAS-102在亚洲人群中的疗效,也直接支持了其在中国等亚洲国家的上市,填补了这些地区晚期结直肠癌后线治疗的空白。事实上,通过摩熵医药提供的‘研发状态表’,我们可以观察到TAS-102在不同国家上市的时间差以及适应症拓展的轨迹,这对于药企规划国际化布局具有极高的参考价值。”
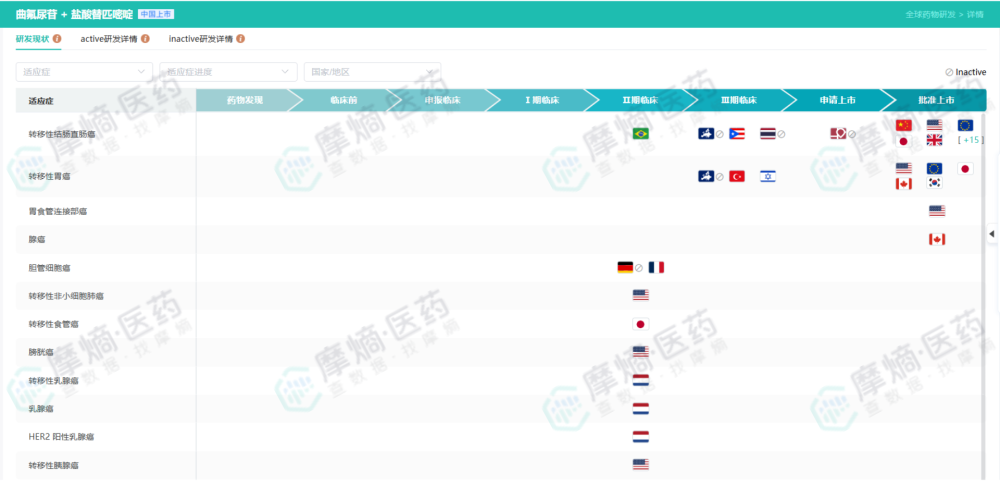
查数据,找摩熵!图源:摩熵医药数据库
七、安全性悖论:中性粒细胞减少是福是祸?
在 TAS-102 的临床应用中,医生们观察到了一个非常反直觉的现象,被称为“安全性悖论”。
TAS-102 最主要的副作用是骨髓抑制,特别是中性粒细胞减少症。在RECOURSE研究中,3级以上中性粒细胞减少的发生率高达38% 。按常理,严重的毒性往往意味着预后不良或需要停药。
然而,对J003和RECOURSE研究的回顾性分析却发现了一个惊人的相关性: 那些在治疗过程中出现了中性粒细胞减少(CINE)的患者,其生存期反而比没有出现该副作用的患者更长 。
深度解析:
- 这提示我们,中性粒细胞减少可能不仅仅是毒性反应,更是药物在体内达到有效生物活性浓度的“生物标志物”。
- 由于个体代谢差异,同样的剂量在不同患者体内产生的血药浓度不同。那些发生骨髓抑制的患者,往往意味着药物在他们体内达到了较高的暴露量,因此对肿瘤的杀伤也更彻底。
临床启示:这一发现彻底改变了临床医生的管理策略。当患者出现中性粒细胞减少时,医生不再轻易恐慌停药,而是通过“减量不停药”或“适度延迟下一周期”的方式,尽力维持治疗。因为他们知道,这看似凶险的指标,其实是疗效的“军功章”。
八、未来展望:超越结直肠癌
TAS-102 的故事并未止步于此。作为一种机制独特的核苷类药物,它的潜力正在被不断挖掘。
1. 胃癌的突破(TAGS研究)
在结直肠癌取得成功后,研究人员迅速将其推向了另一大消化道肿瘤——胃癌。TAGS III期临床研究显示, TAS-102 显著延长了晚期胃癌/胃食管结合部腺癌患者的总生存期(5.7个月 vs 3.6个月,HR=0.69)。这也使其获得了胃癌适应症的批准。
2. 强强联手:TAS-102 + 贝伐珠单抗
最新的SUNLIGHT研究表明,TAS-102与抗血管生成药物 贝伐珠单抗(Bevacizumab)联用,可以产生“1+1>2”的效果 。
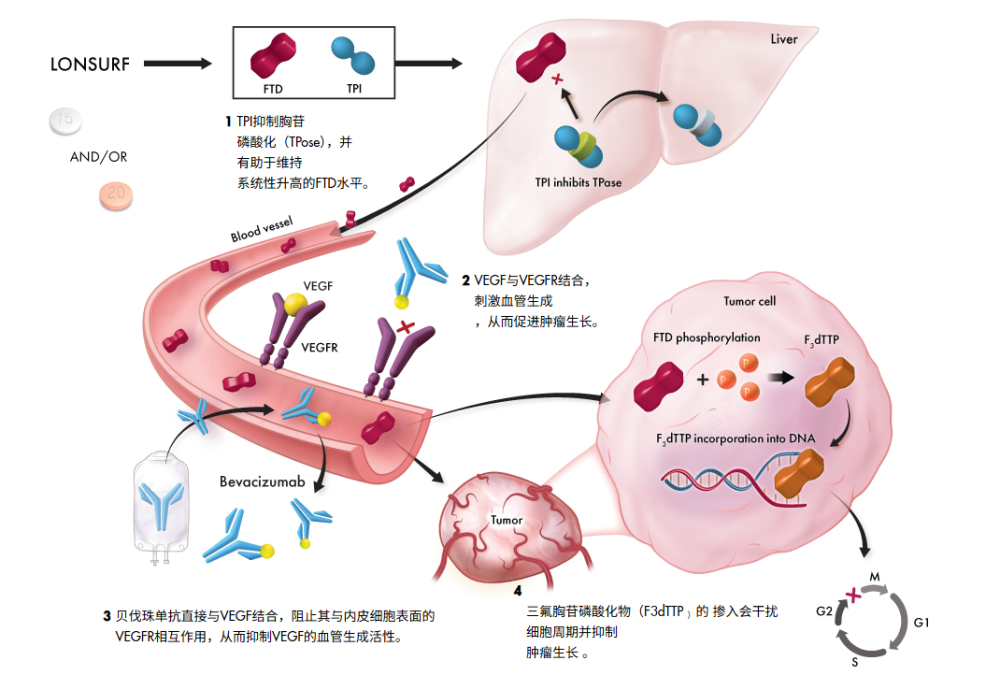
图源:Lonsurf
机制互补:TAS-102直接杀伤肿瘤细胞,贝伐珠单抗切断血管供应,同时TPI本身也有抗血管作用,三者形成了全方位的封锁。
这一组合已被推荐为难治性转移性结直肠癌的新标准治疗。
总结
曲氟尿苷-替匹嘧啶 的研发,绝不仅仅是两个化合物的物理混合。
- 它是对药代动力学(PK)的极致利用;
- 它是对药物作用机制(MOA)的深刻理解;
- 它是转化医学(从临床失败回到实验室再回到临床)的胜利。
对于今天的药物研发人员来说,FTD-TPI 证明了:没有绝对“无用”的分子,只有还没找到正确“打开方式”的药物。 当我们面对看似失败的管线时,或许该问自己一句:是不是它的“替匹嘧啶”还没出现?
往期精彩:
1. 生物技术革命简史:重组DNA、基因泰克与人类基因组计划如何催生千亿美元抗体药物市场
2. 动物模型进化史:揭秘Wistar大鼠、裸鼠与转基因技术,如何成为AI时代前药物研发的“活体显微镜”
3. 新药研发全景图:从分子发现到临床确证,再到上市后监测的挑战
查数据,找摩熵!想要解锁更多药品信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论