


































药物研发,曾经被视为一门基于“试错”的实验艺术。但在信息技术指数级增长的推动下,它正在演变为一门严谨的“计算科学” 。
为什么现在的 AI 能从海量数据中“提取”出药物分子?这不仅仅是 AlphaFold 或 GPT 的单点突破,而是物理载体(硬件)、逻辑表达(软件)与认知模式(算法)三者共生进化的结果 。
从冯·诺依曼架构的瓶颈突破 ,到 GPU/TPU 的算力分化 ;从晦涩的机器码到可视化的数据流编程 ——每一个层级的技术跃迁,都在为模拟生命复杂性打开新的维度。
今天咱来讲讲人类是如何一步步获得“计算”生命的能力。
1. 物理载体的进化
计算能力的物理基础决定了我们能够模拟的生物学复杂度的上限。从宏观的真空管到微观的纳米片,硬件的每一次微缩,都为药物设计打开了新的维度。
1.1 电子开关的微缩史
计算机的本质是状态的切换与存储。这种二元逻辑(0与1)的物理实现经历了四个决定性的历史阶段,每一代技术的更迭都伴随着能效比与计算密度的数量级飞跃。
第一代:热离子真空管的局限(1940-1950年代)
最早的电子计算机,如1946年的ENIAC和IBM 650,依赖于热离子真空管。这些玻璃封装的器件类似于灯泡,利用热电子发射来控制电流。然而,它们的体积庞大、散热量惊人且极易损坏。在药物发现领域,这一时期的算力仅能支持最基础的统计计算,无法触及分子模拟的门槛。
第二代与第三代:固态物理的胜利(1950-1970年代)
1947年晶体管的发明标志着计算进入固态时代。IBM 7090等机型利用晶体管取代了真空管,使得计算机的体积大幅缩小,可靠性显著提升。随后,集成电路(IC)的出现将多个晶体管蚀刻在单一硅片上(如IBM 360/91),这种高密度的集成使得执行早期的量子化学计算(如Hückel分子轨道法)成为可能。
第四代:超大规模集成电路(VLSI)与纳米极限(1980年代至今)
随着光刻技术的进步,我们进入了超大规模集成电路时代。从微米级到纳米级,摩尔定律预言了晶体管密度的指数增长。当前,人类工程学正逼近物理极限。IBM在2021年发布的2纳米(2nm)芯片技术,采用了全环绕栅极(GAA)纳米片架构。2纳米甚至小于人类DNA单链的直径(约2.5纳米)。这种技术能够在指甲盖大小的芯片上容纳500亿个晶体管,相比主流的7纳米技术,其性能提升了45%,能耗降低了75%。
1.2 冯·诺依曼架构
尽管硬件尺寸不断缩小,但主流计算机的逻辑结构——冯·诺依曼体系结构——在过去70年中保持了惊人的稳定性。
架构解析,冯·诺依曼架构的核心在于存储与计算的分离:
(1)中央处理器(CPU):包含控制单元(指挥交通)和运算单元(算术与逻辑操作)。
(2)存储器:同时存储程序指令和数据,处于同一线性地址空间。
(3)输入/输出系统:与外部世界的交互接口。
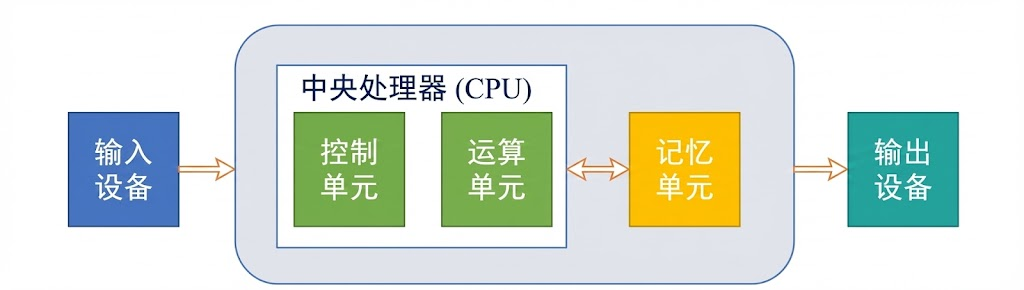
这一架构与生物神经元有着功能上的同构性:神经元的树突接收信号(输入),细胞体进行电位累加与阈值判断(运算/激活),轴突传递信号(输出)。然而,两者存在本质差异。生物大脑的存储与计算是融合的(突触既是连接也是存储),而冯·诺依曼架构存在著名的“冯·诺依曼瓶颈”:由于CPU与存储器之间共享数据总线,指令的读取与数据的传输不能同时进行,导致高速CPU常常处于等待数据的“饥饿”状态。
1.3 算力分化
为了突破瓶颈,特别是在处理深度学习所需的矩阵运算时,计算硬件发生了明显的分化。
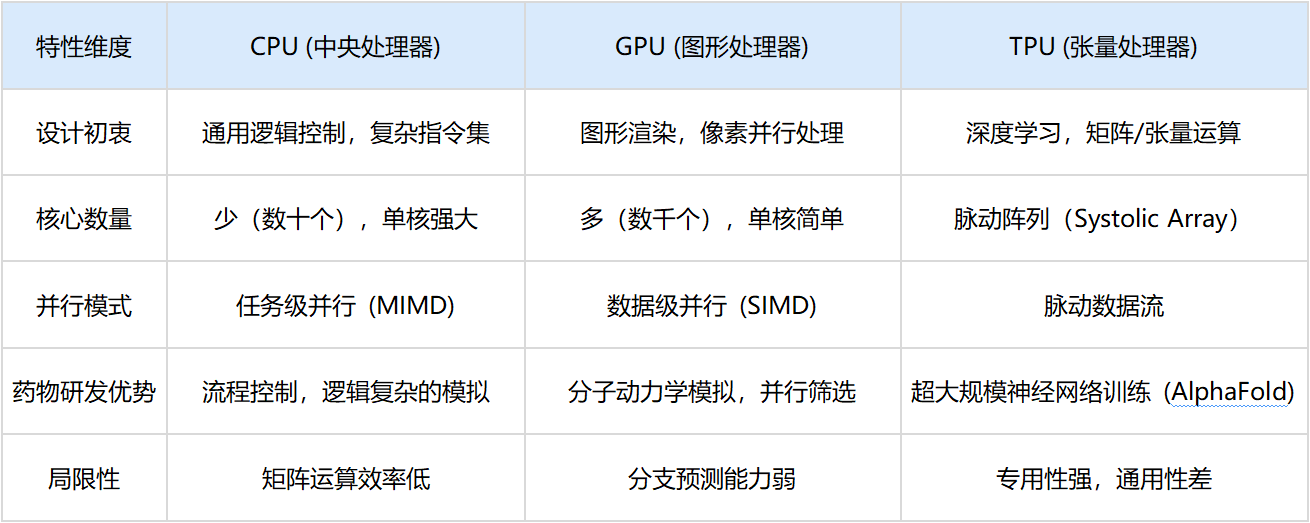
CPU擅长处理复杂的逻辑分支和即时响应,如操作系统的调度。但在药物发现中,面对数百万分子的相似性计算,CPU的串行处理能力显得力不从心 。
GPU最初为游戏渲染设计,但科学家发现,计算屏幕上百万像素的光影变化,与计算蛋白质中百万原子的受力情况,在数学上是高度一致的(都是矩阵乘法)。NVIDIA通过CUDA架构释放了这种能力,使分子动力学模拟(MD)的时间尺度从纳秒级延伸至微秒级 。
Google研发的TPU是硬件演化的最新形态——专用集成电路(ASIC)。TPU抛弃了通用的缓存和控制逻辑,采用了脉动阵列架构。数据像血液流过心脏一样流过芯片,每一个时钟周期都在进行乘加运算,无需频繁访问内存。这种设计专为深度学习中的张量(Tensor)计算优化,使得AlphaFold等巨型模型的训练时间从数月缩短至数天 。
2. 软件逻辑的升维
如果说硬件是躯体,软件则是灵魂。编程语言的演化史,就是一部人类试图降低与机器沟通成本、提高抽象层级的历史。
2.1 编程范式的五代更迭
第一、二代:机器与汇编(底层驱动)
直接操作寄存器与内存地址,效率极高但开发极其痛苦。在药物发现早期,仅用于编写最核心的数学库。
第三代:结构化编程(过程驱动)
C语言、FORTRAN的出现是革命性的。它们引入了“函数”和“逻辑块”的概念。化学家开始能够编写程序来计算分子量、模拟反应动力学。这里的核心思想是分治法:将复杂的药物设计问题分解为一个个子过程。
第四代:面向对象编程(对象驱动)
C++和Java引入了“对象”(Object)的概念。在化学信息学中,这对应着极佳的隐喻:一个“分子”就是一个对象,它拥有属性(原子列表、键列表)和方法(计算LogP、旋转构象)。面向对象编程(OOP)成为了构建大型药物数据库和模拟软件(如ChemOffice)的基石。
第五代:脚本与元语言(数据驱动)
Python、R以及Pipeline Pilot等工具的兴起,标志着编程进入了“胶水语言”时代。化学家不再关心内存管理或指针,而是关注数据流。Python凭借其简洁的语法和庞大的生态库(DeepChem, RDKit, PyTorch),成为了AI药物发现事实上的标准语言。
2.2 可视化数据流
在药物研发的实际场景中,科研人员往往不具备深厚的代码功底。可视化数据流编程语言应运而生,彻底改变了工具的使用方式。
Pipeline Pilot(商业软件)和KNIME(开源软件)是这一范式的代表。它们将复杂的算法封装为一个个“节点”,用户只需将节点通过“管道”(Pipe)连接,即可通过拖拽完成复杂的任务。
- 工作流逻辑:数据(如SMILES列表)从一个节点流入,经过过滤(如Lipinski五规则筛选)、计算(QSAR预测)、转换,最终流出结果。
- 优势:这种“管道化”思维与化学合成的流水线高度契合,极大地促进了跨学科协作,使得生物学家也能利用机器学习模型进行预测。
2.3 数据结构
在计算机中表达一个分子,需要特定的数据结构。
基本类型与复合结构: 计算机底层只有整数、浮点数和字符。但在现代药物计算中,对一个“药物对象”的定义已经远远超越了化学结构本身。除了利用结构体存储原子坐标外,我们还需要挂载更复杂的元数据。例如,在摩熵医药数据库的逻辑中,一个药物实体不仅包含其化学结构(SMILES),还通过指针关联着其“生命周期状态”——包括它在全球各地的专利到期日、临床试验的历史沿革以及注册审批进度。
这种将“微观化学信息”与“宏观情报信息”封装在同一数据结构中的处理方式,让计算机不仅能计算分子的结合能,还能在早期筛选阶段就识别出那些虽然活性极佳、但在专利上已被封锁的“无效”分子。
SMILES与SELFIES:化学语言的进化
- SMILES:通过ASCII字符串线性表达分子图(如苯环表示为c1ccccc1)。它是药物发现中最通用的语言,但对AI不友好,因为随机生成的SMILES往往是非法化学结构(如开环未闭合)。
- SELFIES:一种自引用的鲁棒性表达。在SELFIES中,原本无效的字符串也会被解释为有效的化学结构。这种“100%有效性”对生成式AI至关重要,保证了模型生成的分子在化学上是合理的。
3. 人工智能的认知跃迁
人工智能(AI)在药物发现中的应用,经历了从“教计算机规则”到“让计算机自己学习规则”的根本性转变。
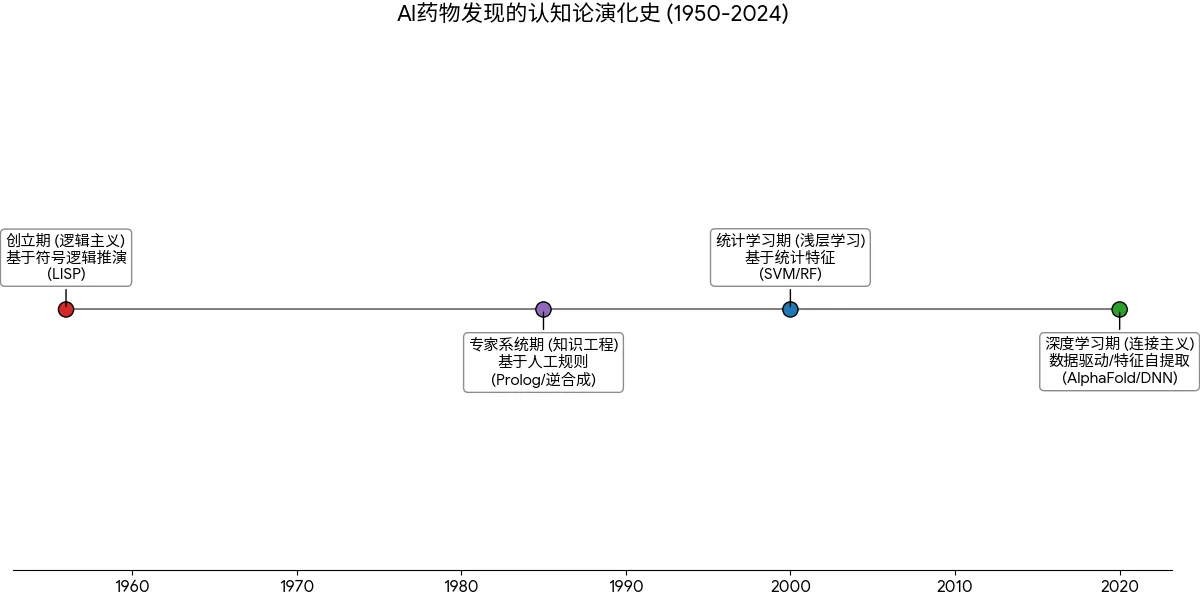
3.1 AI的四个历史时期
创立期(逻辑主义): 1950年代,以LISP语言为代表,试图通过符号逻辑推演智能。虽然提出了神经网络雏形,但受限于算力,主要停留在理论层面。
专家系统期(知识工程): 1980年代,以Prolog语言和E.J. Corey的逆合成分析系统为代表。核心思想是将有机化学家的经验固化为成千上万条“如果-那么”规则。虽然能解决特定问题,但缺乏泛化能力,无法处理未知领域的知识。
统计学习期(浅层学习): 1990-2010年,随机森林(RF)、支持向量机(SVM)等统计模型占据主流。Deep Blue击败人类棋手是算力的胜利,而非学习能力的胜利。
深度学习期(连接主义): 2010年至今。随着AlphaGo和AlphaFold的横空出世,AI进入深度神经网络(DNN)时代。这一阶段的特征是数据驱动:不再人工定义特征(如“氢键供体数量”),而是让网络从海量原始数据中自动提取高维特征。
3.2 深度学习的底层逻辑
现代AI的核心是人工神经网络(ANN),其本质是通用的函数拟合器。
- 神经元与函数的等价性
一个人工神经元就是一个数学函数y = f (w · x + b)。其中 x 是输入向量,w 是权重矩阵,b 是偏置, f 是非线性激活函数(如ReLU或Sigmoid)。无数个这样的函数嵌套、堆叠,形成深度网络,理论上可以逼近任何复杂的非线性映射关系(如从分子结构映射到生物活性)。
- 反向传播与梯度下降
网络“学习”的过程,就是不断调整权重 w 的过程。通过计算预测值与真实值的误差,利用链式法则将误差反向传播回网络,计算每个权重的梯度,并沿梯度方向微调权重。这正是TPU等硬件擅长的高速矩阵运算。
4. 深度神经网络架构
针对不同类型的化学数据(向量、图像、序列、图),深度学习演化出了多种专门的架构,重塑了药物设计的各个环节。
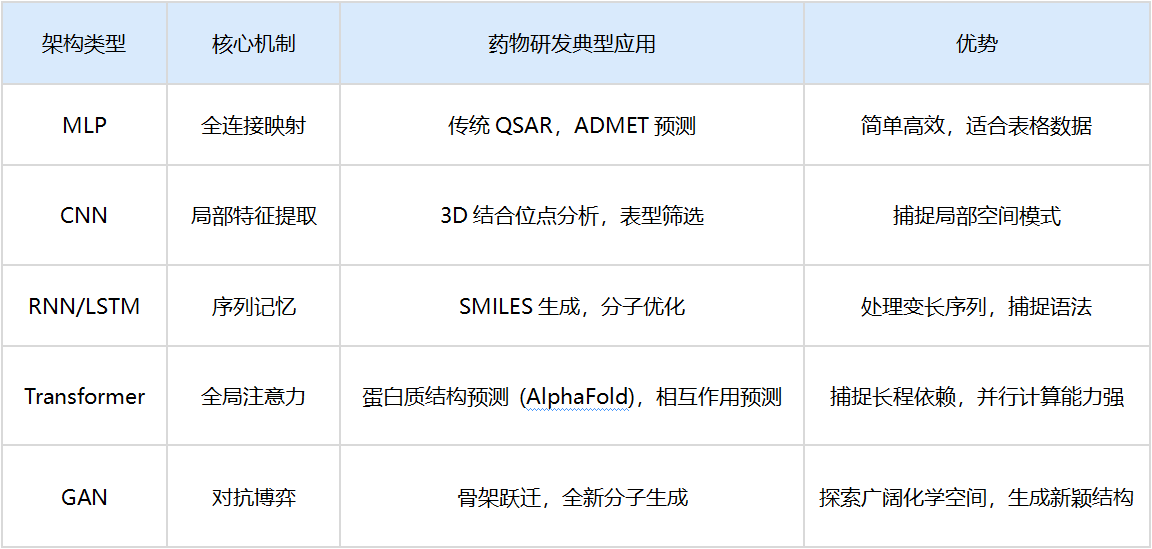
4.1 多层感知器(MLP)
架构:全连接的前馈网络。
应用:QSAR(定量构效关系)。输入分子的理化描述符(如LogP、分子量),输出活性预测。
局限:它丢失了分子的空间拓扑信息,将分子视为“特征的集合”而非结构实体。
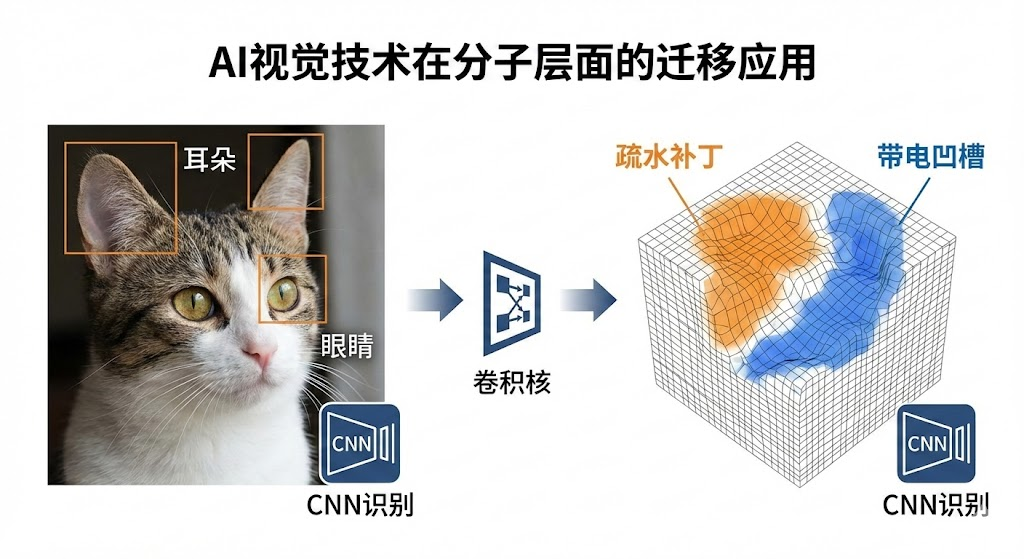
4.2 卷积神经网络(CNN)
原理:利用卷积核(Filter)在数据上滑动,提取局部特征。
药物发现应用:
- 2D CNN:将分子图视为图像处理,或用于分析高内涵筛选(HCS)的细胞图像。
- 3D CNN:将蛋白质的结合口袋视为三维体素网格(Voxel),卷积核在三维空间滑动,识别疏水腔、氢键受体等局部微环境,预测配体结合亲和力。
4.3 循环神经网络(RNN)与LSTM
原理:具有“记忆”的网络,能够处理序列数据。LSTM(长短期记忆网络)引入了门控机制(遗忘门、输入门),解决了长序列中的梯度消失问题。
药物发现应用:处理SMILES字符串。RNN可以将分子设计视为文本生成问题(Next Token Prediction)。通过学习百万级分子的SMILES序列,RNN掌握了化学的“语法”,能够生成全新的、符合化学规则的分子结构。
双向LSTM:同时从前向后和从后向前读取SMILES,捕捉原子间的远距离依赖关系。
4.4 变换器(Transformer)与注意力机制
原理:抛弃了RNN的循环结构,引入自注意力机制(Self-Attention)。这使得网络在处理序列中的某个元素(如氨基酸)时,可以同时关注序列中任意位置的其他元素,无论距离多远。
革命性应用:
- AlphaFold 2/3:利用Evoformer架构(Transformer的变体),同时处理蛋白质序列信息(一维)和残基对的空间几何信息(二维)。注意力机制让模型能够“理解”即便在序列上相距甚远的氨基酸,在折叠后的三维空间中可能是紧密接触的。AlphaFold 3进一步扩展,不仅预测蛋白质,还能预测蛋白质与DNA、RNA及小分子配体的复合物结构。
- 分子BERT:基于Transformer预训练模型,在大规模化学数据库上进行自监督学习,提取深层的分子表征,用于下游的性质预测任务。
4.5 生成对抗网络(GAN)
原理:包含两个对抗的网络——生成器(制造假数据)和判别器(鉴别真假)。两者在博弈中共同进化。
药物发现应用:从头药物设计(De Novo Design)。生成器试图创造出判别器无法区分的“类药分子”。通过引入强化学习奖励(如活性、合成可及性),GAN可以定向生成针对特定靶点的新颖分子结构,突破人类化学家的想象边界。
5. 数据与大模型时代的挑战与机遇
5.1 生物医药大数据的特征
随着高通量筛选(HTS)、基因测序(NGS)和冷冻电镜技术的普及,生物医药数据呈现出爆炸式增长,具备了典型的4V特征:体量大(Volume)、速度快(Velocity)、模态多(Variety)、价值密度低(Value)。这些数据不仅包括结构化的化合物库,还包括非结构化的科学文献、专利、医疗影像等。
5.2 大语言模型(LLM)的跨界融合
2023年以来,以ChatGPT为代表的大语言模型展现了惊人的能力。在药物发现中,LLM不再仅仅是聊天机器人,而是演化为认知引擎:
- 知识抽取:自动阅读数百万篇文献,构建靶点-疾病-药物的知识图谱。
- 代码生成:辅助编写PyTorch代码或KNIME脚本,降低AI使用门槛。
- 多模态推理:未来的模型将是“多模态”的,能够同时理解文本描述(“抑制EGFR突变”)和分子结构数据,实现语义级的药物搜索与设计。
6. 结语
曾经,研发一款新药是一场耗资十亿美元、耗时十年的豪赌;如今,TPU 与生成式 AI 正在将这场豪赌变成可控的工程 。 当“试错”的成本被转移到云端,当无效的分子在合成之前就被剔除,我们正在见证制药工业效率的指数级跃迁 。这场跨越 70 年的认知变革,最终指向了一个朴素的目标:让好药来得更快,更便宜。
扩展阅读:
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论