

































1.4.1 本周全球TOP10创新药研发进展
(1)FDA批准全球首款SAA细胞疗法上市
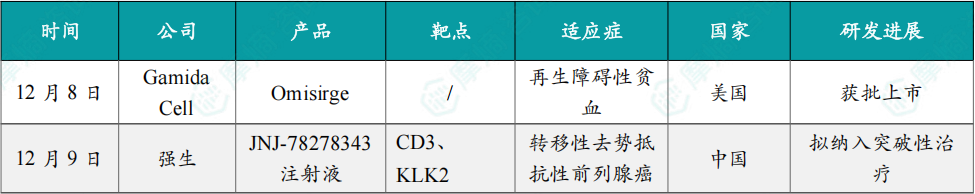
12月8日,美国FDA正式批准Gamida Cell公司开发的同种异体脐带血细胞疗法 Omisirge®(通用名:omidubicel-onlv)用于治疗重型再生障碍性贫血(SAA)。
这是全球首个获批用于SAA的细胞疗法产品,标志着这一罕见且危及生命的骨髓衰竭性疾病迎来革命性治疗突破。
值得注意的是,Omisirge® 最初于2023年获FDA批准,用于12岁及以上血液系统恶性肿瘤患者在接受清髓性预处理后的脐带血移植,以缩短中性粒细胞恢复时间并降低感染风险。此次新增适应症覆盖6岁及以上、缺乏匹配捐赠者的SAA患者,使其成为全球首个兼具恶性与非恶性血液病治疗能力的“双适应症”细胞疗法,进一步拓展了其临床价值与应用广度。
(2)强生双抗新药JNJ-78278343注射液拟纳入突破性疗法
12月9日,CDE官网显示,强生的1类新药 JNJ-78278343注射液 拟纳入突破性治疗品种,适应症为用于接受过雄激素受体(AR)通路抑制剂和紫杉烷类化疗治疗的转移性去势抵抗性前列腺癌(mCRPC)成人患者。公开数据显示,该药是全球首个且目前唯一进入Ⅲ期临床阶段的KLK2靶向药物。
JNJ-78278343(Pasritamig)是强生开发的一款潜在 first-in-class 皮下注射TCE(T 细胞桥接器)双抗,可同时靶向CD3和KLK2。其中,KLK2中文全称为人激肽释放酶2,是一种在前列腺癌细胞表面表达的蛋白,但在正常组织中表达非常有限。
目前,强生正在中国、美国、日本、法国等多个国家和地区开展一项国际多中心Ⅲ期临床(CTR20254394),以评估 JNJ-78278343 联合最佳支持治疗用于转移性去势抵抗性前列腺癌患者的有效性和安全性,研究预计将于2028年5月完成。
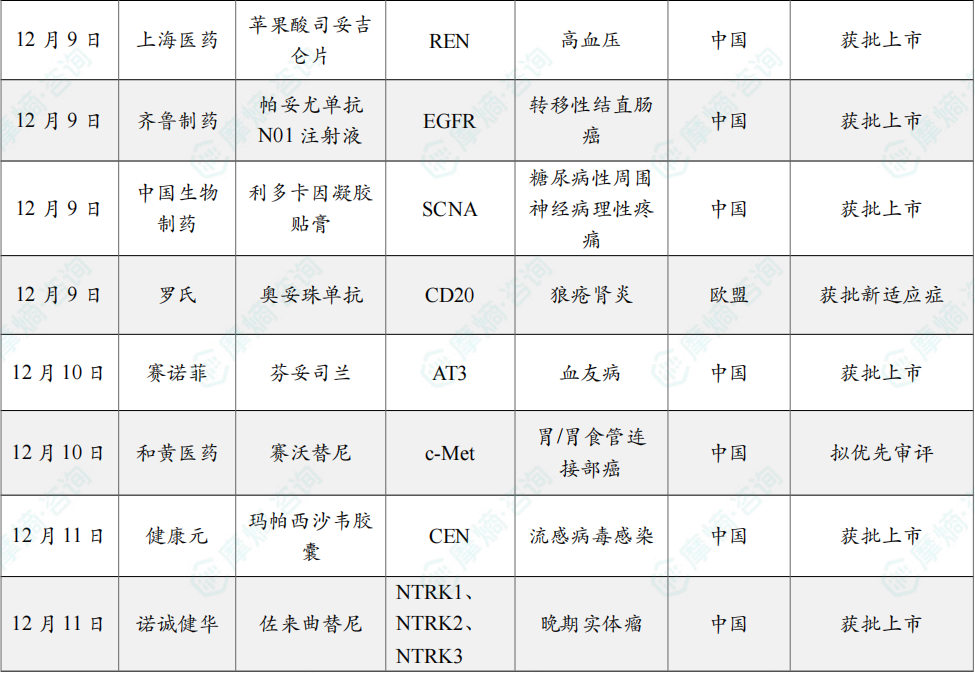
(3)上海医药高血压1类新药苹果酸司妥吉仑片获批上市
12月9日,国家药品监督管理局(NMPA)官网显示,上海医药的1类新药 苹果酸司妥吉仑片(SPH3127)新药上市申请(NDA)已获批准,用于治疗高血压。
司妥吉仑是新一代口服非肽类小分子肾素抑制剂,通过对肾素的直接抑制,拮抗由肾素-血管紧张素-醛固酮系统(RAAS)过度激活造成的血压上升,适用于原发性高血压的治疗,由上海医药和日本田边三菱制药株式会社合作研发。
2023年5月,该药III期临床试验确证性研究(SPH3127-301 第二阶段)主要研究终点结果达到方案预设的非劣标准。临床试验结果显示:司妥吉仑100mg/次,口服,每日一次,连续用药 12 周(试验方案规定的用药期)治疗原发性轻、中度高血压是安全、有效的。司妥吉仑可为原发性高血压患者提供一种新的治疗选择。
(4)齐鲁制药的帕尼单抗生物类似药获批上市
12月9日,国家药品监督管理局(NMPA)官网显示,齐鲁制药的 帕尼单抗生物类似药(QL1203)获批上市,用于联合mFOLFOX一线治疗RAS野生型转移性结直肠癌(mCRC)患者。
原研帕尼单抗是武田与安进合作开发的一款靶向表皮生长因子受体(EGFR)的单克隆抗体,暂时未在中国获批上市,齐鲁制药 QL1203 的获批因此具有重要的临床意义。
EGFR是重要的原癌基因,帕尼单抗 可以针对EGFR异常细胞信号通路,抑制肿瘤生长。齐鲁开展了一项III期注册性临床研究,旨在评估 QL1203 联合 mFOLFOX6 对比安慰剂联合 mFOLFOX6 一线治疗RAS野生型的mCRC患者的有效性和安全性。
研究结果显示,在既往未接受过抗EGFR疗法的RAS/BRAF野生型mCRC患者中,QL1203 联合 mFOLFOX6 与安慰剂联合 mFOLFOX6 相比,显著改善了PFS并获得更高的ORR。QL1203 联合 mFOLFOX6 耐受性与安全性良好。QL1203 在与化疗联合一线治疗结直肠癌患者的研究中展现出良好的安全性和可耐受性,安全谱与原研药物相似。
(5)中国生物制药全球首个糖尿病神经痛贴剂获批上市
12月9日,中国生物制药宣布,旗下泰德制药自主研发的 利多卡因凝胶贴膏(商品名:得百宁®)新适应症获批上市,用于缓解糖尿病性周围神经病理性疼痛(DPNP)治疗,成为全球首个、国内唯一获批 DPNP 治疗的贴剂药物。
作为全球首个 DPNP 适应症外用贴剂,其采用独特骨架型水凝胶交联技术,含 700 mg 高剂量利多卡因,搭配尿素、丙二醇等促透剂,37% 基质含水量保障药物缓慢释放,贴敷 12 小时内即可实现持续镇痛。
临床数据显示,该产品无口服药常见的全身副作用,皮肤过敏发生率极低,也不影响肝肾功能及代谢指标,因不良反应停药率显著低于口服药组。此外,利多卡因凝胶贴膏也可与口服药联用,成为其“增效减量搭档”。
(6)罗氏CD20单抗奥妥珠单抗获欧盟批准新适应症
12月9日,罗氏宣布欧盟委员会批准 Gazyva/Gazyvaro(奥妥珠单抗)联合 吗替麦考酚酯(MMF)用于治疗成人活动性III级或IV级(可合并V级)狼疮肾炎。
此次批准基于II期NOBILITY和III期REGENCY研究的积极结果。发表于《新英格兰医学杂志》的REGENCY关键数据显示,Gazyva/Gazyvaro 联合标准治疗(MMF+糖皮质激素)组46.4%的患者达到完全肾脏缓解,而标准治疗单用组为33.1%。研究同时观察到皮质类固醇用量显著减少、蛋白尿应答改善,以及补体水平升高、抗dsDNA下降等炎症与疾病活动度标志的临床意义改善。安全性与其血液肿瘤适应症中已确立的特征一致。
Gazyva/Gazyvaro 是目前唯一在随机III期研究中证明能带来完全肾脏缓解获益的抗CD20抗体。2025年10月,美国FDA也批准了 Gazyva/Gazyvaro 用于接受标准治疗的成人活动性狼疮肾炎。
(7)赛诺菲创新血友病疗法在国内获批上市
12月10日,赛诺菲宣布其RNAi疗法 芬妥司兰(Fitusiran,中文商品名:赛菲因,英文商品名:Qfitlia)获得国家药监局(NMPA)批准上市,用于患有以下疾病的12岁及以上儿童和成人患者的常规预防治疗,以防止出血或降低出血发作频率:存在或不存在凝血因子VIII抑制物的重型A型血友病(先天性凝血因子VIII缺乏,FVIII<1%)或存在或不存在凝血因子IX抑制物抑制物的重型B型血友病(先天性凝血因子IX缺乏,FIX<1%)患者。
该产品是血友病领域首款RNAi疗法,一年最少只需注射6次。芬妥司兰 是Alnylam Pharmaceuticals开发的一款靶向抗凝血酶III(AT III)的RNAi疗法。2014年1月,赛诺菲以7亿美元收购了该公司12%的股份,获得了包括 芬妥司兰 在内的4款RNAi疗法的权益。目前,赛诺菲仅拥有 芬妥司兰 和 Revusiran 的权益。
(8)和黄医药的赛沃替尼胃癌适应症拟优先审批
12月10日,CDE官网显示,和黄医药的 赛沃替尼 新适应症拟优先审批,用于治疗经过至少2种系统化疗失败的MET基因扩增的局部晚期或转移性胃癌或胃食道连接部腺癌成人患者。
赛沃替尼是和黄医药开发的一种口服的高选择性c-Met抑制剂,可阻断因突变 (例如外显子14跳跃突变或其他点突变) 、基因扩增或蛋白质过表达而导致的MET受体酪氨酸激酶信号通路的异常激活。
2011年12月,阿斯利康与和黄医药达成合作协议,前者主导 赛沃替尼 在中国的开发工作,阿斯利康则主导海外开发工作。此外,和黄医药负责 赛沃替尼 在中国的上市许可、生产和供应,而阿斯利康则负责实现赛沃替尼在中国乃至全球范围内的商业化。
(9)健康元1类抗流感新药玛帕西沙韦胶囊获批上市
12月11日,健康元宣布,1类创新药 玛帕西沙韦(TG-1000胶囊)获NMPA批准上市,适用于既往健康的12岁及以上青少年和成人单纯性甲型和乙型流感患者的治疗,不包括存在流感相关并发症高风险的患者。
玛帕西沙韦 为创新抗流感1类新药,是一种新型帽依赖性核酸内切酶抑制剂,最早由太景医药研发。2023年3月,太景医药授予健康元该药的知识产权许可以独家研发、生产、销售、许诺销售 玛帕西沙韦。
III期临床试验结果表明,玛帕西沙韦 对甲型和乙型流感病毒感染患者均展现良好的疗效,对乙型流感的疗效优于同类药物。在III期临床研究中,玛帕西沙韦 组与安慰剂组在所有流感症状缓解的中位时间分别为60.9小时和87.9小时,较安慰剂组缩短27小时,显示达到主要疗效指标并具有统计学差异。
(10)诺诚健华抗癌新药佐来曲替尼获批上市
12月11日,NMPA官网显示,诺诚健华1类新药 佐来曲替尼 在国内获批上市,用于治疗携带NTRK融合基因的晚期实体瘤成人和青少年(12~18周岁)患者。
佐来曲替尼(曾用名:卓乐替尼;研发代号:ICP723)是诺诚健华自主研发的泛TRK抑制剂,可以克服第一代TRK抑制剂的获得性耐药。
目前,诺诚健华已经完成了针对NTRK融合基因阳性的晚期实体瘤成人和青少年患者的关键注册临床试验(登记号:NCT05745623),该试验的主要疗效终点是IRC评估的ORR。在临床研究中,佐来曲替尼 展示了卓越的有效性和安全性,IRC评估的ORR为 89.1%,DCR为96.4%,24个月PFS率为77.4%,24个月OS率为90.8%。此外,佐来曲替尼 针对儿童患者(2周岁≤年龄<12周岁)的注册临床试验正在进行中。
1.4.2 本周全球TOP10积极/失败临床结果
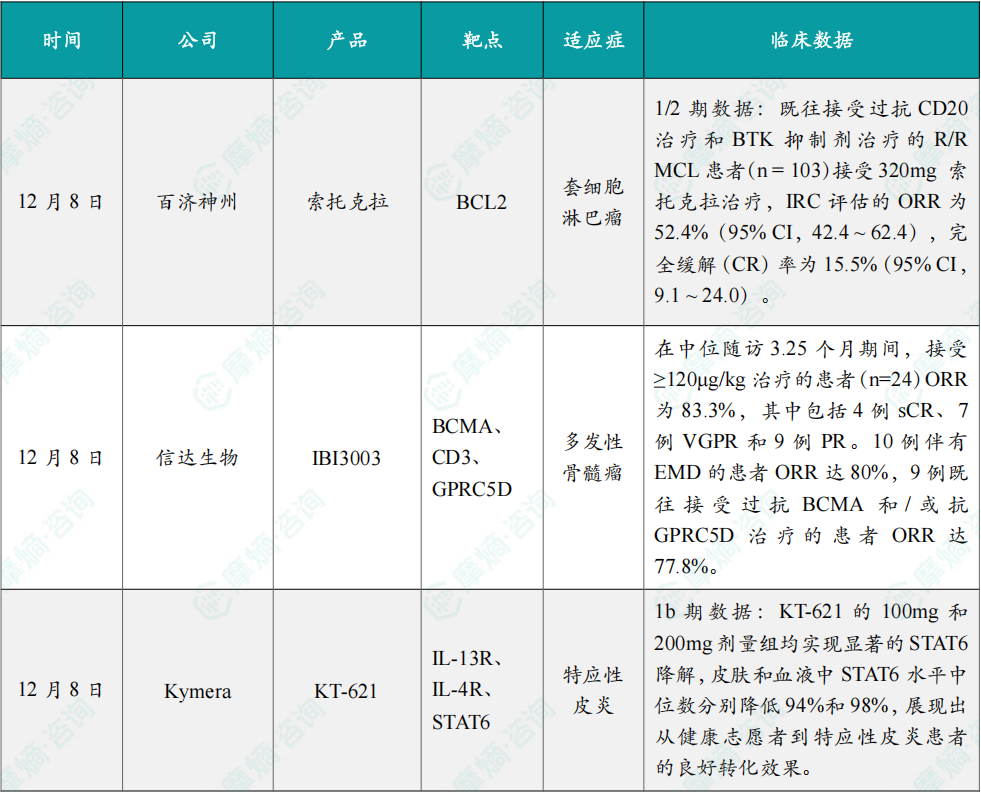
(1)百济神州公布BCL2抑制剂索托克拉1/2期临床积极结果
12月8日,百济神州宣布,新一代在研BCL2抑制剂 索托克拉 五项临床研究的最新数据表明,无论是单药还是联合用药,其在多种B细胞恶性肿瘤的治疗中均展现出显著的临床获益,具备作为基石用药的潜力:在用于治疗既往接受过多线治疗的复发或难治性(R/R)套细胞淋巴瘤(MCL)患者中,索托克拉 展现出持久缓解的效果。
美国血液学会(ASH)2025年会上公布了上述研究的具体数据。在这项全球、多中心、单臂、开放性1/2期研究BGB-11417-201(NCT05471843)中,既往接受过抗CD20治疗和BTK抑制剂治疗的R/R MCL患者(n=103)接受320mg 索托克拉治疗,IRC评估的ORR为52.4%(95% CI,42.4 ~ 62.4),完全缓解(CR)率为15.5%(95% CI,9.1 ~ 24.0)。基于此数据,索托克拉 有望成为美国首款获批用于治疗R/R MCL的BCL2抑制剂。
(2)信达生物公布三特异性抗体首次人体试验初步数据,针对多发性骨髓瘤
12月8日,信达生物在2025年美国血液学会年会(ASH)上以口头报告形式首次公布其自主开发的抗GPRC5D/BCMA/CD3三特异性抗体 IBI3003 用于复发或难治性多发性骨髓瘤(R/R MM)患者的首次人体试验的初步数据。
IBI3003 展示出良好的耐受性和可控的安全性特征,尽管当前随访时间尚短,IBI3003 已经显现出令人鼓舞的初步疗效信号,特别在伴有髓外病变或既往接受抗BCMA和GPRC5D单一或双重靶向治疗的高危患者中展现了积极的疗效。
具体而言,在中位随访3.25个月期间,接受≥120μg/kg治疗的患者(n=24)ORR为83.3%,其中包括4例sCR、7例VGPR和9例PR。10例伴有EMD的患者ORR达80%,9例既往接受过抗BCMA和/或抗GPRC5D治疗的患者ORR达77.8%。
(3)Kymera公布KT-621 1b期临床积极数据
12月8日,Kymera Therapeutics宣布,其first-in-class STAT6 PROTAC药物 KT-621 在中重度特应性皮炎(AD)患者中开展的BroADen Ib期临床试验取得积极结果。KT-621 的BroADen Ib期试验是一项开放标签、单臂研究,共纳入22例中重度特应性皮炎患者(分两个连续队列入组)。其中10例受试者每日口服100mg KT-621,12例受试者每日口服200mg KT-621,治疗周期为28天,随后进入14天随访期。
试验结果显示,KT-621 的100mg和200mg剂量组均实现显著的STAT6降解,皮肤和血液中STAT6水平中位数分别降低94%和98%,展现出从健康志愿者到特应性皮炎患者的良好转化效果。同时,KT-621 可显著降低血液中多种2型疾病相关生物标志物水平,减少皮损部位核心2型炎症及特应性皮炎相关基因集表达。
安全性方面,KT-621 表现优异,研究期间无严重不良事件、无治疗相关不良事件发生,未报告结膜炎病例,生命体征、实验室检查及心电图均无临床相关异常变化。
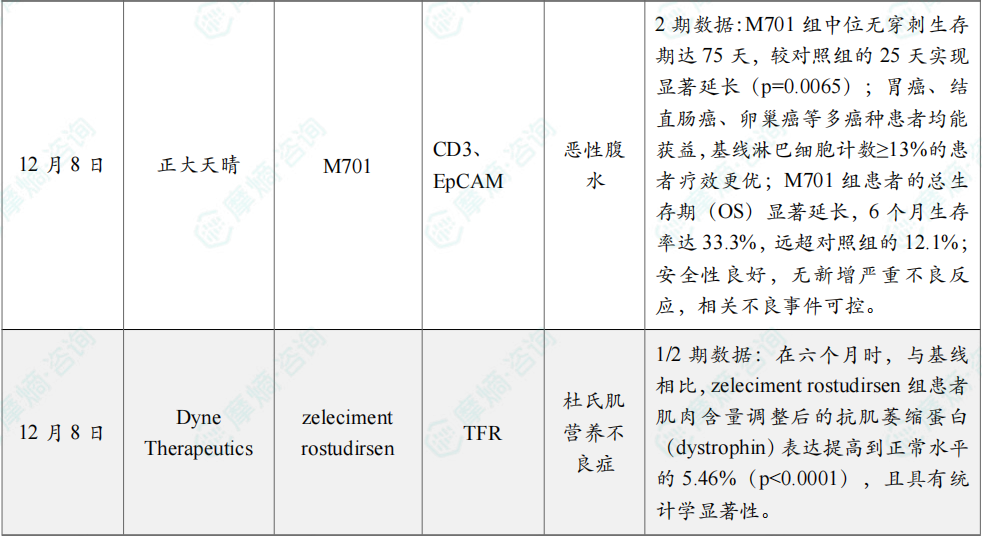
(4)正大天晴引进双抗2期临床成果登《EHO》,治疗恶性腹水
12月8日,正大天晴宣布,从友芝友生物引进的1类创新药 M701 治疗恶性腹水(MA)的Ⅱ期临床研究结果已于近日正式发表于《Experimental Hematology & Oncology》(EHO,IF 13.5),无穿刺生存期和总生存期较对照组显著延长。
M701 是国内首个自主开发并进入临床试验阶段的CD3/EpCAM双特异性抗体,拟用于肿瘤引起的恶性胸水(MPE)和恶性腹水(MA)的治疗。此次公布Ⅱ期临床研究共纳入84例患者。结果显示:M701 组中位无穿刺生存期达75天,较对照组的25天实现显著延长(p=0.0065);胃癌、结直肠癌、卵巢癌等多癌种患者均能获益,基线淋巴细胞计数≥13%的患者疗效更优;M701 组患者的总生存期(OS)显著延长,6个月生存率达33.3%,远超对照组的12.1%;安全性良好,无新增严重不良反应,相关不良事件可控。
(5)Dyne Therapeutics在研疗法zeleciment rostudirsen 1/2期临床试验取得积极结果
12月8日,Dyne Therapeutics宣布,其在研疗法 zeleciment rostudirsen 在1/2期DELIVER试验的注册性扩展队列(REC)中,用于适合外显子51跳跃治疗的杜氏肌营养不良症(DMD)患者时取得了积极顶线结果。
Dyne表示其预计在2026年第二季度向美国FDA递交加速批准的上市申请目前进展良好。分析显示,REC达到试验主要终点,显示在六个月时,与基线相比,zeleciment rostudirsen 组患者肌肉含量调整后的抗肌萎缩蛋白(dystrophin)表达提高到正常水平的5.46%(p<0.0001),且具有统计学显著性。此结果重现了此前在注册剂量观察到的抗肌萎缩蛋白相较基线增加7倍的变化。
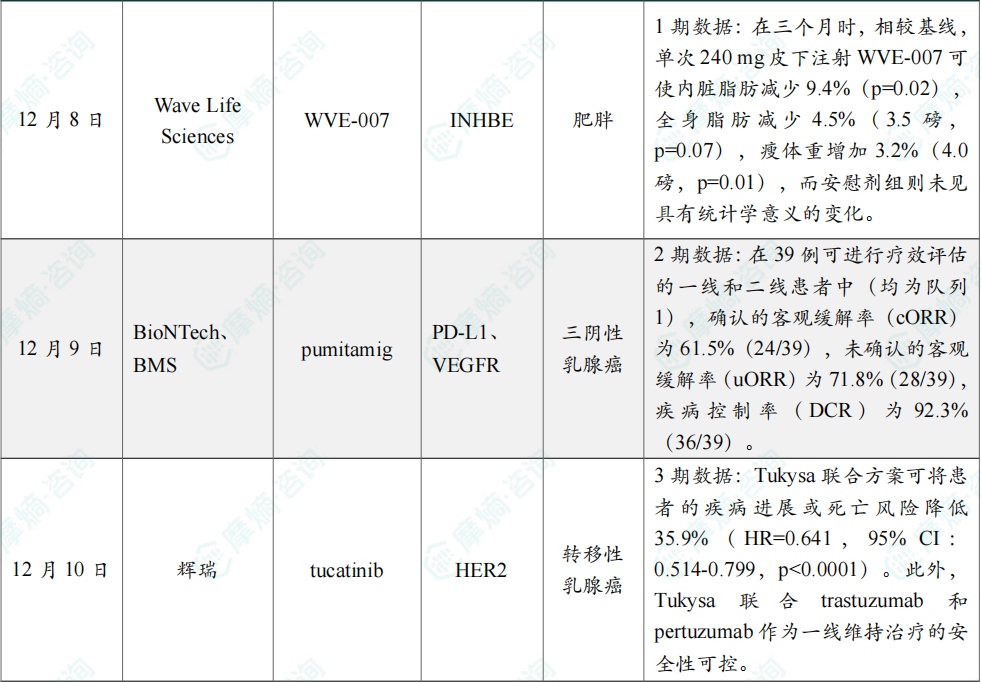
(6)Wave Life Sciences减肥药1期临床中期数据公布
12月8日,Wave Life Sciences公布了正在进行的全球首个Ⅰ期人体试验INLIGHT中最低治疗剂量组的积极中期数据。在此次中期评估中,单次给予240 mg WVE-007 后三个月,受试者身体成分出现改善,表现为总体脂肪和内脏脂肪质量减少、瘦体重增加;同时展现出良好的安全性,且血清Activin E水平出现持久下降,支持潜在的一年一次或一年两次给药方案。
数据显示,在三个月时,相较基线,单次240 mg皮下注射 WVE-007 可使内脏脂肪减少9.4%(p=0.02),全身脂肪减少4.5%(3.5磅,p=0.07),瘦体重增加3.2%(4.0磅,p=0.01),而安慰剂组则未见具有统计学意义的变化。
试验结果还显示,受试者血清中的Activin E持续且显著受到抑制,有望带来身体组成的持续改善、进一步减脂并维持肌肉量。整体来看,该药物安全性良好,仅观察到轻度治疗相关不良事件,实验室检测指标(包括血脂和肝功能)未见具有临床意义的异常变化。
(7)BioNTech和BMS公布pumitamig 2期临床中期数据
12月9日,BioNTech和百时美施贵宝(Bristol Myers Squibb)公布了一项全球随机2期临床试验的中期数据。该试验评估了双抗 pumitamig 联合化疗对局部晚期或转移性三阴性乳腺癌(TNBC)患者的疗效与安全性,无论患者的PD-L1表达水平为何。
截至2025年10月1日的中期分析,共纳入74例接受pumitamig联合标准治疗化疗的局部晚期/转移性TNBC患者。试验主要结果如下:在39例可进行疗效评估的一线和二线患者中(均为队列1),确认的客观缓解率(cORR)为61.5%(24/39),未确认的客观缓解率(uORR)为71.8%(28/39),疾病控制率(DCR)为92.3%(36/39)。不同剂量水平、PD-L1表达水平以及治疗线别的患者亚群均显示出令人鼓舞的疗效,且观察到更高药物剂量与患者更高的缓解率相关。15 mg/kg剂量组患者的uORR为63.2%;20 mg/kg为80.0%;PD-L1表达水平CPS≥10组患者的uORR为70.6%;CPS<10为70.6%;一线治疗患者的uORR为76.5%;二线患者则为68.2%。
(8)辉瑞公布HER2小分子抑制剂3期临床积极数据
12月10日,辉瑞公布其在人表皮生长因子受体2(HER2)阳性转移性乳腺癌(MBC)患者中开展的3期HER2CLIMB-05研究的积极结果。该研究评估患者在接受化疗诱导治疗后,接受小分子 Tukysa(tucatinib)或安慰剂,与标准一线维持治疗方案(trastuzumab联合pertuzumab)联用时的疗效与安全性。
主要终点分析显示,在研究者评估下,Tukysa联合方案可将患者的疾病进展或死亡风险降低35.9%(HR=0.641,95% CI:0.514-0.799,p<0.0001)。此外,Tukysa 联合 trastuzumab 和 pertuzumab 作为一线维持治疗的安全性可控。
Tucatinib 是一种口服酪氨酸激酶抑制剂,对HER2具有高度特异性,但对同属人表皮生长因子受体家族的EGFR没有明显抑制作用。目前,该药已获FDA批准两项适应症。
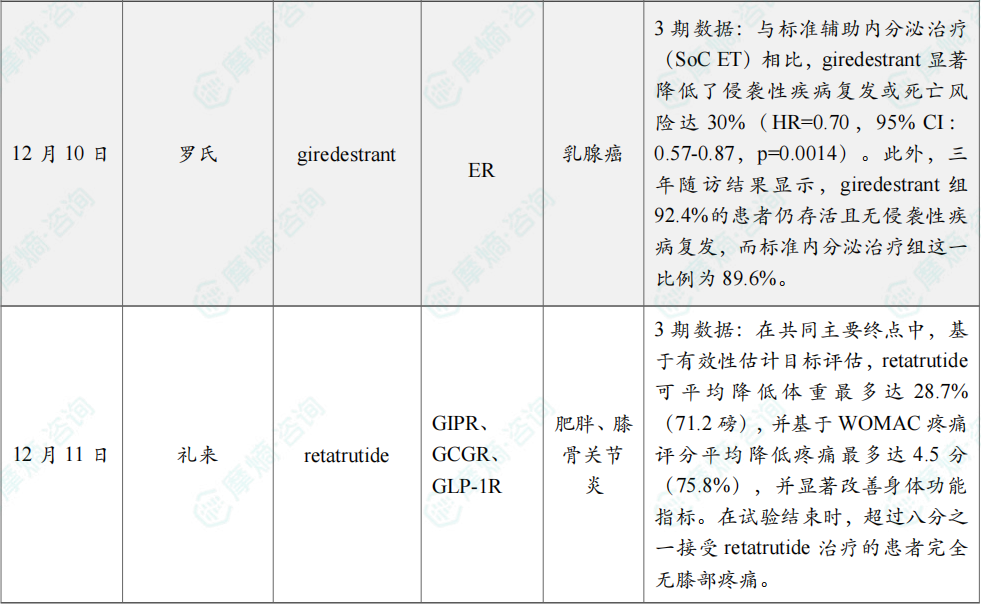
(9)罗氏公布口服SERD 3期临床积极数据,用于治疗乳腺癌
12月10日,罗氏公布了3期lidERA Breast Cancer研究的积极结果,该研究评估在研口服新一代选择性雌激素受体降解剂(SERD)giredestrant 在雌激素受体(ER)阳性、HER2阴性的早期乳腺癌患者中的疗效与安全性。
预设期中分析结果显示,giredestrant 组患者在主要终点无侵袭性疾病生存期(iDFS)上获得改善。与标准辅助内分泌治疗(SoC ET)相比,giredestrant 显著降低了侵袭性疾病复发或死亡风险达30%(HR=0.70,95% CI:0.57-0.87,p=0.0014)。
此外,三年随访结果显示,giredestrant 组92.4%的患者仍存活且无侵袭性疾病复发,而标准内分泌治疗组这一比例为89.6%。iDFS获益在所有具有临床相关性的亚组中均保持一致。
(10)礼来三靶点减重疗法retatrutide 3期积极结果公布
12月11日,礼来(Eli Lilly and Company)宣布,其TRIUMPH-4临床3期试验取得积极的主要研究结果。该试验评估其在研、每周一次、潜在“first-in-class”葡萄糖依赖性促胰岛素多肽(GIP)、GLP-1和胰高血糖素(glucagon)受体靶向三重激动剂 retatrutide,在患有肥胖或超重且合并膝骨关节炎、但无糖尿病的成年人中,作为健康饮食与运动的辅助治疗方案时的安全性和有效性。
分析显示,患者在接受12 mg retatrutide 治疗68周后,平均减重达28.7%。在这项全球注册性试验中,84.0%的参与者基线身体质量指数(BMI)≥35 kg/m²。试验中,retatrutide 的两种最高研究剂量(9 mg和12 mg)均达成所有主要及关键次要终点,在68周时基于有效性估计目标(efficacy estimand)和治疗方案估计目标(treatment-regimen estimand)均实现显著体重下降,并在疼痛及身体功能方面取得改善。在共同主要终点中,基于有效性估计目标评估,retatrutide 可平均降低体重最多达28.7%(71.2磅),并基于WOMAC疼痛评分平均降低疼痛最多达4.5分(75.8%),并显著改善身体功能指标。在试验结束时,超过八分之一接受 retatrutide 治疗的患者完全无膝部疼痛。
同期事件:
1. 2025年第50周12.08-12.14国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2025年第50周12.08-12.14国内仿制药/生物类似物申报/审批数据分析
3. 2025年第50周12.08-12.14国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2025.12.08-2025.12.14) },如需查看或下载完整版报告,可点击!
扩展阅读:
1. 2024年第50周12.09-12.15全球创新药研发概览
2. 2024年第51周12.16-12.22全球创新药研发概览
3. 2024年第52周12.23-12.29全球创新药研发概览
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!














 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论