


































在人类与疾病漫长的博弈史中,药物研发无疑是最锋利的武器之一。而要让这把武器精准命中目标,就必须找到体内的“靶点”(Target)。所谓的药物靶点,通俗来讲,就是药物在体内发挥作用的那个“着力点”,通常是细胞上的生物大分子,如蛋白质或核酸。药物分子通过与这些靶点进行特异性的结合,改变它们的构象或功能,从而引发一系列生理生化反应,最终达到治疗疾病的目的。
在现代药物研发的浩瀚星空中,虽然新兴靶点层出不穷,但有几类“老牌劲旅”凭借其在生理活动中的核心地位和极高的成药性,占据了现有市售药物靶点的半壁江山,被称为药物发现中的“经典靶点”。它们分别是:G蛋白偶联受体(GPCRs)、离子通道、酶、以及转运体和核受体。深入理解这些靶点的作用机制,不仅是掌握药理学基础的关键,更是洞察新药研发逻辑的必经之路。
一、G蛋白偶联受体(GPCRs)
G蛋白偶联受体(GPCR)在信号转导中发挥着重要作用。这类膜结合蛋白家族能为细胞提供机体维持正常功能所需的信息,是通信网络的主要部分。它是人体内最大的膜蛋白受体家族,也是目前最成功的药物靶点类别,大约有三分之一的现代药物都是通过作用于GPCRs来发挥疗效的。
1. 结构与机制
GPCRs的结构非常有特色,它像一条长蛇,在细胞膜上来回穿梭了七次,因此也被称为“七跨膜受体”。它的胞外部分负责“接收信号”,即结合细胞外的激素、神经递质或其他信号分子(配体);而它的胞内尾部则连接着一个关键的合作伙伴——G蛋白(鸟苷酸结合蛋白)。
当细胞外的信号分子(比如肾上腺素)作为“访客”按响了这个“门铃”(与GPCR胞外侧结合)时,GPCR的整体结构会发生微妙的扭曲变化。这个变化就像一个开关,激活了胞内的G蛋白。被激活的G蛋白就像一个短跑运动员,迅速离开受体,在细胞膜内侧跑去激活下游的效应器(通常是酶或离子通道)。这些效应器随后会产生大量的“第二信使”分子(如cAMP、钙离子等),将信号在细胞内逐级放大,最终引发细胞的特定生理反应,如心跳加快、肌肉收缩或腺体分泌。
这个过程就像一场精密的接力赛:细胞外信号是第一棒,GPCR是交接棒的区域,G蛋白是第二棒,而后续的第二信使则是将比赛推向高潮的第三棒。
2. 药物的作用方式
针对GPCRs的药物设计主要有两种思路:
- 激动剂(Agonist):这类药物扮演的是“按铃人”的角色。它们模仿人体内天然的信号分子,与GPCR结合并激活它,从而引发后续的信号级联反应。例如,用于治疗哮喘的沙丁胺醇就是一种β2肾上腺素受体激动剂,它能激活气道平滑肌上的受体,使气道舒张。
- 拮抗剂(Antagonist):这类药物则更像是“堵锁眼”的恶作剧者。它们能与GPCR结合,但不会激活受体,而是占据了天然信号分子的结合位点,阻止了“门铃”被按响。例如,广泛用于治疗高血压和心脏病的“β-受体阻滞剂”(如普萘洛尔),就是通过阻断肾上腺素与β受体的结合,从而降低心率和血压。
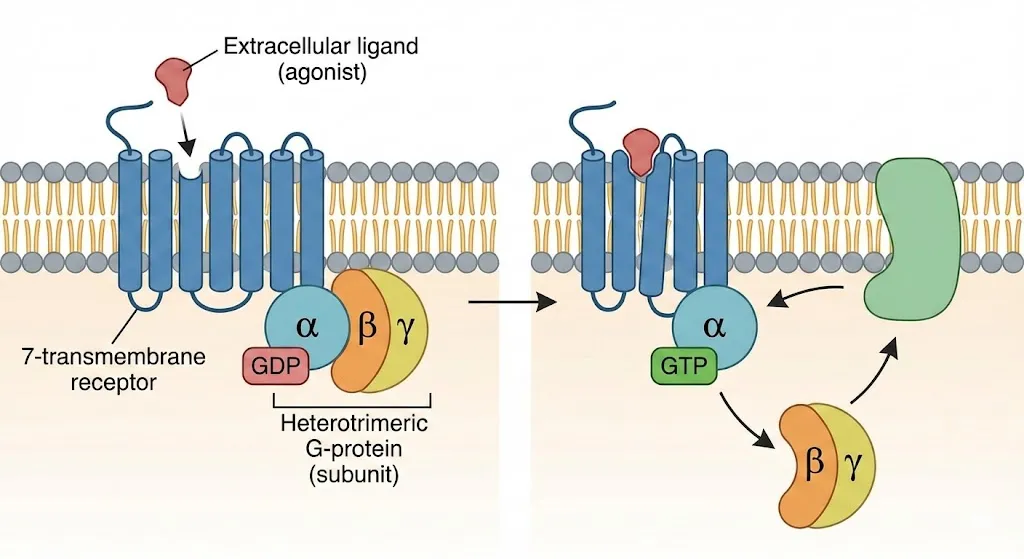
图:G蛋白偶联受体(GPCR)的激活机制。左侧显示静息状态下,受体未与配体结合,G蛋白以异源三聚体形式存在并结合GDP。右侧显示激动剂结合后,受体构象改变,G蛋白α亚基上的GDP被GTP取代,导致α亚基与βγ亚基解离,进而激活下游信号通路。
二、离子通道
离子通道(ion channel)作为一类跨膜蛋白组件,在调节离子生物屏障跨膜流动的过程中发挥着重要作用。细胞内外的离子浓度差异(如钠、钾、钙、氯离子)是维持细胞正常生理功能,特别是神经冲动传导和肌肉收缩的基础。离子通道就是镶嵌在细胞膜上的一类蛋白质复合物,它们在细胞膜上形成了一个个亲水的孔道,充当着“边防检查站”的角色,严格控制着特定离子进出细胞。
1. 类型:电压门控与配体门控
离子通道的开闭不是随意的,而是受到严格调控的。根据“开关”方式的不同,主要分为两大类:
- 电压门控离子通道:它们的开关受细胞膜两侧电位差变化的控制。当神经元发生去极化时,这些通道会迅速打开,允许离子(如钠离子)涌入,产生动作电位。这是神经信号快速传导的基础。
- 配体门控离子通道:它们的开关受特定化学信号分子(配体)的控制。当神经递质(如乙酰胆碱、谷氨酸、GABA等)与通道上的特定位点结合时,通道就会打开或关闭。这类通道通常位于突触后膜,负责将化学信号转换为电信号。
2. 药物的作用方式
作用于离子通道的药物,其核心策略是调节通道的开闭状态,从而改变细胞的兴奋性。
- 通道阻滞剂(Blockers): 这类药物就像是堵住通道口的“塞子”,物理性地阻止离子通过。例如,局部麻醉药(如利多卡因)就是一种钠通道阻滞剂。它们阻断了神经纤维上的电压门控钠通道,使得疼痛信号无法传导到大脑,从而产生麻醉作用。
- 通道调节剂(Modulators): 这类药物的作用更为精妙,它们通常结合在通道的辅助位点(变构位点)上,改变通道对天然配体的敏感性或改变通道开放的概率。例如,抗焦虑药苯二氮䓬类药物(如安定)就是作用于GABA-A受体(一种氯离子通道)的变构调节剂。它们本身不能打开通道,但能增强抑制性神经递质GABA的作用,使氯离子通道更容易打开,导致神经元超极化,兴奋性降低,从而产生镇静、抗焦虑的效果。
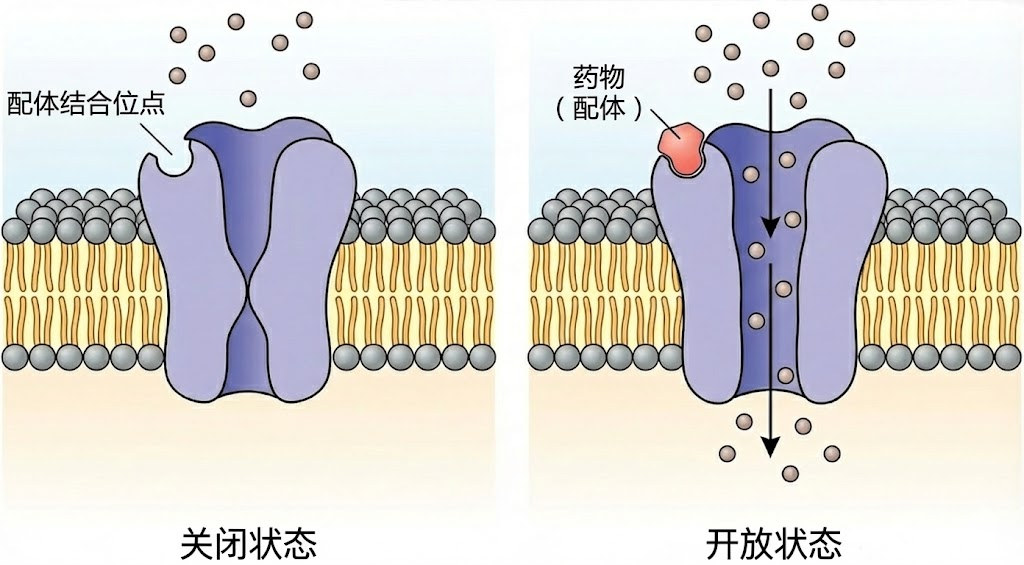
图:配体门控离子通道的开闭机制。左侧显示通道处于关闭状态,离子无法通过。右侧显示当药物分子作为配体结合到通道上的特定位点后,引发通道构象变化,中心孔道打开,允许特定离子(小球所示)顺浓度梯度穿过细胞膜。
三、酶
酶是一类天然的蛋白催化剂,可以帮助完成生命所需的化学转化。它们催化着体内几乎所有的生物化学反应,从食物的消化代谢到DNA的复制修复,无一不需要酶的参与。许多疾病的发生都与特定酶的活性异常(过高或过低)有关,因此,酶自然成为了药物研发的重要靶点。
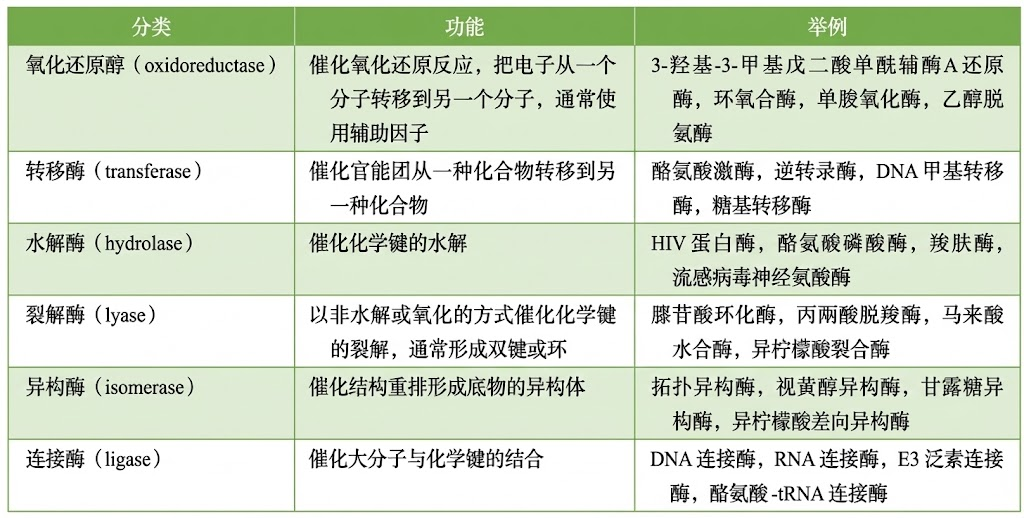
六大种类酶的典型实例
1. 机制
酶的作用机制通常用“锁与钥匙”模型来描述。酶分子上有一个特定的区域称为“活性中心”,其形状和电荷分布与特定的底物分子高度互补。底物像钥匙一样插入活性中心的锁孔中,形成酶-底物复合物。在活性中心,底物的化学键变得不稳定,从而更容易发生化学反应生成产物。
2. 药物的作用方式
大多数作用于酶的药物都是酶抑制剂,它们通过降低酶的活性来发挥疗效。抑制剂的作用方式多种多样,其中最常见的是竞争性抑制。
竞争性抑制剂(Competitive Inhibitors):这类药物在结构上与天然底物非常相似,它们就像一把“假钥匙”,能插入酶的活性中心,但无法引发后续的化学反应。由于药物分子占据了活性中心,天然底物就无法进入,从而抑制了酶的催化活性。这种抑制是可逆的,增加底物的浓度可以竞争性地置换出药物分子,恢复酶的活性。
- 经典案例:著名的他汀类降脂药(如阿托伐他汀)就是HMG-CoA还原酶的竞争性抑制剂。HMG-CoA还原酶是胆固醇合成途径中的关键限速酶,他汀类药物通过抑制该酶的活性,有效地降低了体内胆固醇的合成。
非竞争性抑制剂(Non-competitive Inhibitors): 这类药物结合在酶活性中心以外的位点(变构位点)。它们的结合会导致酶的构象发生变化,使得活性中心虽然没有被占据,但已经变形,无法有效地催化底物。这种抑制作用无法通过增加底物浓度来消除(尽管药物与酶的结合本身在化学上可能是可逆的)。
不可逆抑制剂(Irreversible Inhibitors): 通过共价连接至目标酶的活性位点, 阻断天然底物的进入,进而使酶失活。
- 经典案例:阿司匹林就是一种不可逆抑制剂。它通过乙酰化环氧化酶(COX)活性中心的一个关键丝氨酸残基,永久性地使该酶失活,从而阻断了前列腺素的合成,发挥解热、镇痛、抗炎和抗血小板聚集的作用。由于血小板没有细胞核,无法重新合成COX酶,因此阿司匹林的抗血小板作用可以持续整个血小板的生命周期(约7-10天)。
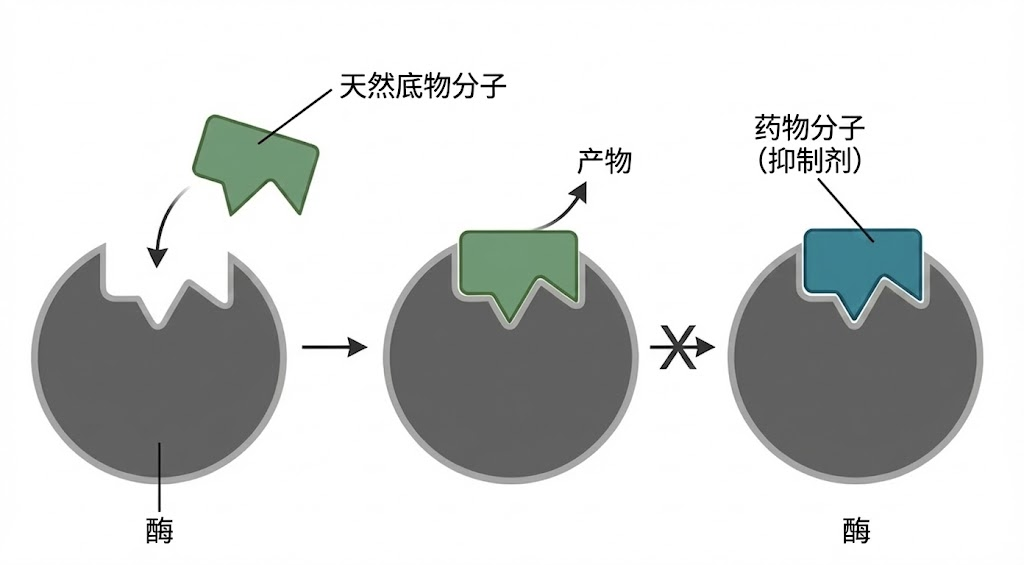
图:酶的竞争性抑制机制。左图显示天然底物与酶的活性中心特异性结合,并被催化生成产物。右图显示一种与天然底物结构相似的药物分子(抑制剂)竞争性地结合到酶的活性中心,占据了位置,从而阻止了天然底物的结合和后续反应。
四、转运体
细胞膜是一道脂质屏障,许多水溶性的小分子物质(如葡萄糖、氨基酸、神经递质等)和离子无法自由通过。膜转运蛋白(membrane transport protein,也称为转运蛋白或转运体)所主导的跨膜运输是大多数小分子跨越生物屏障的主要方式。转运体相当于就是细胞膜上负责这些物质跨膜运输的“物流队”。它们通过构象变化,将特定的物质从膜的一侧转运到另一侧。
1. 类型:被动运输与主动运输
被动转运体(Facilitators):它们协助物质顺浓度梯度进行跨膜运输,不需要消耗能量。例如,葡萄糖转运体(GLUTs)负责将血液中的葡萄糖运送到细胞内。
主动转运体(Pumps):它们能逆浓度梯度运输物质,需要消耗能量(通常来自ATP水解)。例如,钠钾泵(Na+/K+-ATPase)不断地将细胞内的钠离子泵出,将细胞外的钾离子泵入,维持细胞内外离子的浓度差和静息电位。
2. 药物的作用方式:“截流”与“反向利用”
药物可以通过抑制转运体的功能来发挥疗效,也可以利用转运体进入细胞。
抑制转运体:许多药物通过抑制神经递质的再摄取转运体来发挥作用。
- 经典案例1:选择性5-羟色胺再摄取抑制剂(SSRIs),如氟西汀(百忧解),就是通过抑制突触前膜上的5-羟色胺转运体(SERT),阻断了5-羟色胺从突触间隙回到突触前神经元的过程,从而提高了突触间隙中5-羟色胺的浓度,增强了神经信号的传递,发挥抗抑郁作用。
- 经典案例2:利尿剂(如呋塞米)通过抑制肾小管上的Na+-K+-2Cl-共转运体,减少了钠离子和氯离子的重吸收,从而增加尿量,降低血压和水肿。
利用转运体:一些药物分子设计成能被特定的转运体识别和转运,从而提高药物的口服吸收率或靶向性进入特定细胞。例如,帕金森病治疗药物左旋多巴(L-DOPA)就是利用氨基酸转运体穿过血脑屏障进入脑内,然后转化为多巴胺发挥作用的。
五、核受体
与前面提到的那些位于细胞膜上、反应迅速的受体不同,核受体(Nuclear Receptors)是一类位于细胞内(细胞质或细胞核)的转录因子。它们通常与脂溶性的信号分子(如类固醇激素、甲状腺激素、维生素D等)结合后,直接进入细胞核,结合到特定的DNA序列上,调控特定基因的转录和表达。
1. 机制:慢而持久的基因调控
由于涉及基因转录和蛋白质合成的过程,核受体介导的生理效应通常比较缓慢(数小时到数天),但持续时间较长。例如,糖皮质激素通过结合其核受体,调控多种炎症相关基因的表达,发挥强大的抗炎和免疫抑制作用。
2. 药物的作用方式:激动剂与拮抗剂
作用于核受体的药物同样分为激动剂和拮抗剂。
- 激动剂:模仿天然激素的作用,促进特定基因的表达。例如,用于激素替代疗法的雌激素和孕激素,就是分别作用于雌激素受体(ER)和孕激素受体(PR)的激动剂。
- 拮抗剂:阻断天然激素与受体的结合,抑制特定基因的表达。例如,用于治疗乳腺癌的他莫昔芬,就是一种雌激素受体的选择性调节剂(SERM),在乳腺组织中表现为拮抗剂,阻断雌激素对癌细胞生长的刺激作用;而在骨骼和子宫中则表现为激动剂,有助于维持骨密度。
结语
GPCRs、离子通道、酶、以及转运体和核受体构成了现代药物研发的基石。它们在漫长的进化过程中,被赋予了调控生命活动最核心、最基本的职能。针对这些经典靶点的药物研发,不仅积累了海量的科学数据和丰富的实践经验,也为我们理解人体生理病理机制提供了最直接的窗口。
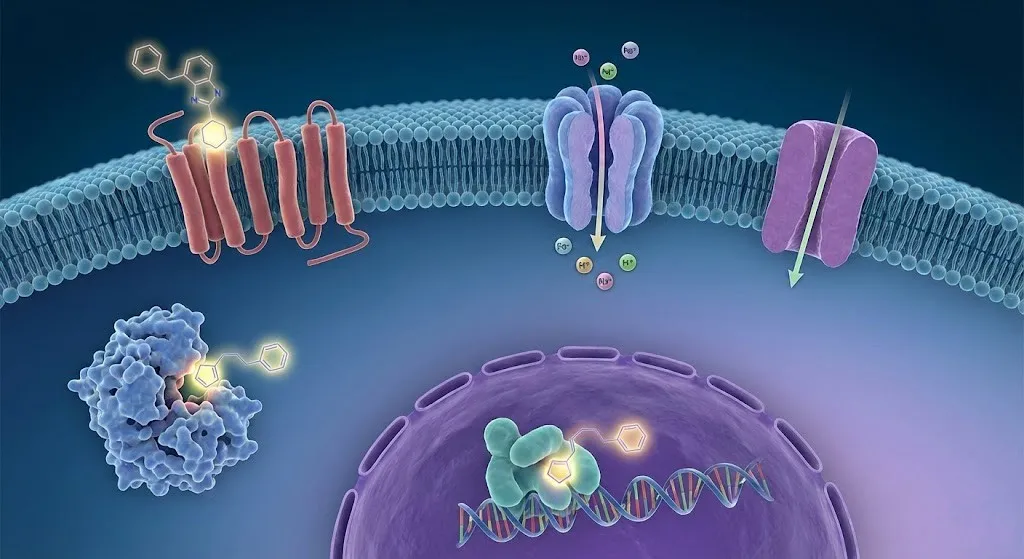
尽管蛋白质-蛋白质相互作用(PPI)、核酸(DNA/RNA)等新兴靶点潜力巨大,但经典靶点(如GPCRs、激酶)凭借其成熟的机制与成药性,仍将在药物研发领域占据不可替代的主导地位。对于研发人员而言,不仅要深耕这些经典靶点的创新机制(如变构调节),更要掌握将“靶点”转化为“药物”的关键技能。
这意味着我们必须从单纯的靶点认知,拓展到对化合物构效关系及构性关系的精准把控——因为结构上的毫厘之差,往往决定了活性的千里之别。为了帮助大家更好地掌握这一技能,下一期,我们来了解下药物化学知识在新药发现与开发中的重要作用~
扩展阅读:
1. 探秘生物药:从重组DNA到细胞移植,解锁疾病治疗全新密码
3. 干货 | 药物筛选五次技术迭代:从表型到AI,虚拟筛选战术图谱大揭秘
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论