


































1.4.1 本周全球TOP10创新药研发进展
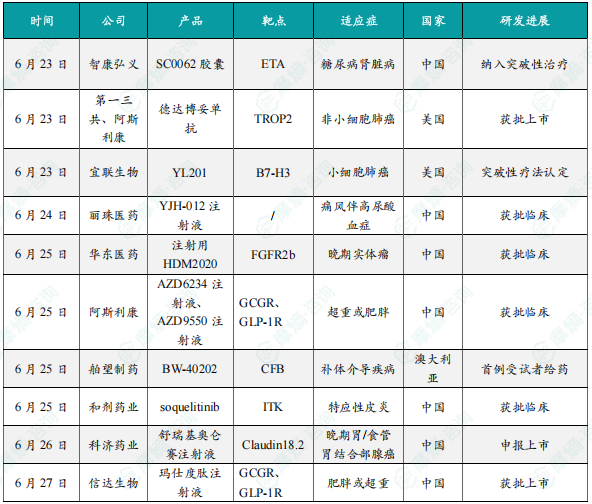
(1)智康弘义小分子肾病新药SC0062胶囊被纳入突破性治疗品种
6月23日,智康弘义宣布其核心管线之一的高选择性内皮素受体A(ETA)小分子拮抗剂SC0062胶囊通过中国国家药品监督管理局(NMPA)药品审评中心(CDE)评审并完成相应公示程序,再次纳入突破性治疗品种名单,用于治疗伴有白蛋白尿的糖尿病肾脏病(DKD)。此前,SC0062已被CDE纳入突破性治疗品种,用于治疗伴有蛋白尿的IgA肾病,目前正在进行3期临床中。本次再获突破性疗法认定标志着SC0062在多种肾脏疾病中的突出临床价值得到进一步认可,也彰显了其成为肾病领域新一代广谱治疗药物的潜力。SC0062是一款针对慢性肾脏病进行全新分子设计、对内皮素受体A(ETA)具有独特高选择性的小分子拮抗剂,1期健康人研究和2-SUCCEED 2期临床试验DKD及先前完成的IgA肾病两个队列的研究结果充分显示,SC0062已具备“best-in-class”特质。
(2)第一三共和阿斯利康的德达博妥单抗肺癌适应症获FDA批准上市
6月23日,第一三共和阿斯利康联合宣布德达博妥单抗(Dato-DXd,商品名:Datroway)在美国获批新适应症,用于治疗既往接受过EGFR靶向药物和铂类化疗治疗的局部晚期或转移性EGFR突变非小细胞肺癌(NSCLC)成人患者。这是FDA批准的首个可用于治疗肺癌的TROP2 ADC药物。德达博妥单抗最初由第一三共开发,2020年7月,第一三共与阿斯利康达成一项超60亿美元的合作,在全球(日本除外)联合开发和商业化德达博妥单抗,这也是继 HER2 ADC德曲妥珠单抗之后,双方在ADC领域达成的第2项合作。
(3)宜联生物的靶向B7-H3的ADC药物YL201获FDA突破性疗法认定
6月23日,宜联生物医药宣布,其自主研发的靶向B7-H3的抗体偶联药物(ADC)YL201获得美国食品药品监督管理局(FDA)突破性疗法认定(Breakthrough Therapy Designation),用于治疗小细胞肺癌(SCLC)。这是全球首款获此认定的B7-H3 ADC药物,标志着中国创新药在实体瘤治疗领域取得重大突破。B7-H3是一种在多种恶性肿瘤中高表达、但在正常组织中表达有限的免疫调节蛋白,尤其在肺癌、鼻咽癌等实体瘤中过表达,与疾病进展和不良预后密切相关。其特性使其成为开发ADC药物的理想靶点:YL201通过靶向B7-H3,将细胞毒素精准递送至肿瘤细胞,减少对正常组织的损伤;除小细胞肺癌外,该药物在鼻咽癌、肺淋巴上皮瘤样癌(LELC)等适应症中均展现显著疗效。随着FDA突破性疗法认定的获批,YL201有望成为首个获批的B7-H3靶向药物,为小细胞肺癌患者提供后线治疗新标准。宜联生物计划于2025年内启动FDA上市申请前会议(pre-BLA),加速推进全球商业化进程。
(4)丽珠医药1类新药YJH-012注射液获批临床
6月24日,中国国家药监局药品审评中心(CDE)官网公示,丽珠医药申报的1类新药YJH-012注射液获批临床,拟开发治疗痛风。公开资料显示,这是一款基于小干扰RNA(siRNA)技术开发的创新药,有望为患者提供疗效更好、安全性更高的长效痛风治疗方案。YJH-012针对痛风伴高尿酸血症,区别于传统药物抑制酶活性以阻断尿酸合成,实现了从基因层面源头长效抑制尿酸生成的机制突破,有望为临床提供更长效、更安全、更彻底的解决方案。YJH-012的核心机制是利用小干扰RNA(siRNA)技术,将经过化学修饰的双链结构分子,通过特定的肝靶向递送系统GalNAc,精准递送至尿酸合成的主要场所——肝脏。在肝细胞内,siRNA分子与RNA诱导沉默复合体(RISC)结合。RISC中的解旋酶分离siRNA双链,释放其引导链。该引导链精确识别并结合靶蛋白对应的信使RNA(mRNA),并引导RISC中的剪切酶将其切割降解。被破坏的mRNA无法再作为模板合成目标蛋白,从而在源头上显著降低了关键致病蛋白的生成量,阻断了尿酸生成通路。
(5)华东医药注射用HDM2020获批临床,用于治疗晚期实体瘤
6月25日,华东医药宣布全资子公司中美华东申报的注射用HDM2020临床试验申请获得中国国家药监局药品审评中心(CDE)批准,适应症为晚期实体瘤。注射用HDM2020是由中美华东研发的1类生物新药,是一款靶向成纤维细胞生长因子受体2b(Fibroblast Growth Factor Receptor 2b,FGFR2b)的新型抗体偶联药物(ADC),可特异性结合表达人FGFR2b的肿瘤细胞并通过向胞内释放毒素载荷,发挥肿瘤杀伤作用。临床前研究已证明HDM2020在靶点阳性的胃癌、鳞状非小细胞肺癌(sq-NSCLC)等药效模型中显示出强大的抗肿瘤活性,具有良好的成药性和安全性。
(6)阿斯利康减重新药AZD6234注射液和AZD9550注射液联合疗法在中国获批临床
6月25日,CDE官网显示,阿斯利康的AZD6234注射液和AZD9550注射液获批临床,联合用于在至少有一种肥胖相关合并症的超重或者肥胖成年人中的长期体重管理。AZD6234为每周一次皮下注射的长效胰淀素受体激动剂,该产品对降钙素受体活性具有选择性,阿斯利康拟开发该产品作为肠促胰岛素不耐受肥胖患者的替代治疗方案。在1期剂量递增研究中,AZD6234显示出疗效和良好的耐受性,单次2.7mg剂量后,全球队列和日本人群队列的平均体重分别下降1.7%和3.8%。该产品正在国际范围内进行2b期临床研究,用于治疗肥胖/超重。AZD9550是一种每周一次的合成GLP-1R/GCGR双肽激动剂,具有胰高血糖素和GLP-1R激动剂的优化比例,正在国际范围内开展针对肥胖症的2期临床研究。在临床前研究中,该产品可导致肥胖小鼠体重的剂量依赖性降低。
(7)舶望制药在研药物BW-40202完成临床1期首例健康受试者给药
6月25日,舶望制药宣布,其在研小干扰RNA(siRNA)药物BW-40202完成临床I期首例健康受试者给药,BW-40202是靶向补体因子B(CFB)的用于治疗补体介导疾病的药物,该里程碑在澳大利亚临床研究中心达成。此外,BW-40202已获得中国国家药品监督管理局(NMPA)的新药临床试验(IND)批准,将在中国临床研究中心同步推进。BW-40202是一种靶向肝脏中CFB mRNA的siRNA药物, 通过RNA干扰(RNAi)机制,抑制CFB mRNA的表达,从而降低血清CFB蛋白水平并抑制补体替代途径(CAP)活性。在临床前研究中,BW-40202表现出优异的纯度和稳定性,能够显著降低血清CFB蛋白水平,有效抑制CAP活性,并具有长效性和良好的安全性。该项首次人体试验将评估BW-40202在健康受试人群的安全性、药代动力学及生物标志物变化。临床前研究数据支持该疗法在治疗补体介导疾病方面具有创新治疗的潜力。
(8)和剂药业小分子新药soquelitinib在中国获批1b/2期临床试验
6月25日,和剂药业宣布,中国国家药品监督管理局药品审评中心(CDE)已批准其在中国启动soquelitinib(CPI-818)治疗特应性皮炎(AD)患者的1b/2期临床试验的申请。Soquelitinib是处于临床阶段的具有高选择性的口服小分子ITK(白细胞介素-2诱导的T细胞激酶)抑制剂,与目前只能抑制单一或有限细胞因子的注射生物疗法相比,soquelitinib作为口服药物,能阻断参与炎症过程的多种细胞因子,有望显著改善特应性皮炎的治疗现状。最新美国临床试验数据显示soquelitinib的ITK抑制疗法将为特应性皮炎患者提供一种更安全、有效且便捷的全新治疗选择。和剂药业拥有soquelitinib的大中华权益,该公司正在开发soquelitinib的多种适应症,包括这次获批的针对中重度AD患者的1b/2期临床试验。
(9)科济药业新药舒瑞基奥仑赛注射液申报上市
6月26日,科济药业宣布,中国国家药品监督管理局(NMPA)已受理舒瑞基奥仑赛注射液(产品编号:CT041)的新药上市申请(NDA),用于治疗Claudin18.2表达阳性、至少二线治疗失败的晚期胃/食管胃结合部腺癌(G/GEJA)患者。此外,科济药业正积极拓展探索舒瑞基奥仑赛注射液在癌症早线及围术期治疗中的应用,包括一项正在进行的用于胰腺癌辅助治疗的Ib期注册临床试验,及一项正在进行的用于G/GEJA患者术后辅助治疗后巩固治疗的研究者发起的临床试验。
(10)信达生物的玛仕度肽注射液获批上市,针对成人肥胖或超重
6月27日,中国国家药监局(NMPA)宣布批准信达生物申报的胰高血糖素(GCG)/胰高血糖素样肽-1(GLP-1)双受体激动剂玛仕度肽注射液上市,用于成人肥胖或超重患者的长期体重控制。该药品适用于在控制饮食和增加体力活动基础上对成人患者的长期体重控制,初始体重指数(BMI)为:BMI≥28 kg/㎡(肥胖);或BMI≥24 kg/㎡(超重),并伴有至少一种体重相关的合并症(例如高血糖、高血压、血脂异常、脂肪肝、阻塞性睡眠呼吸暂停综合征等)。玛仕度肽是是信达生物与礼来(Eli Lilly and Company)共同推进的一款GCG/GLP-1双受体激动减重药物,其具有独特的双激动作用机制,在激动GLP-1受体抑制食欲的基础上,同时激活GCG受体,促进脂肪燃烧,降低内脏脂肪含量,进一步增强玛仕度肽减重效果。
1.4.2 本周全球TOP10积极/失败临床结果
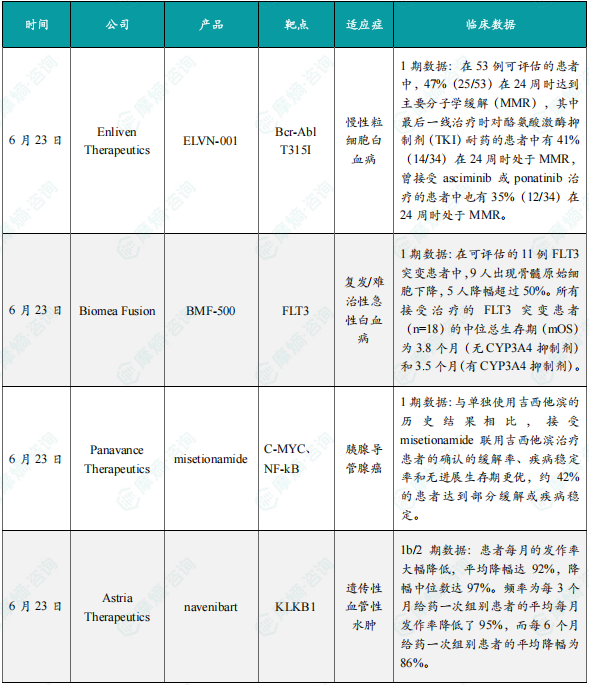

(1)Enliven Therapeutics公司公布ELVN-001 1期临床试验新数据
6月23日,Enliven Therapeutics公司公布其小分子激酶抑制剂ELVN-001治疗慢性粒细胞白血病(CML)患者的1期临床试验的新数据。ELVN-001是一种强效、高选择性、潜在“best-in-class”的小分子激酶抑制剂,专门针对CML患者的致癌驱动因子BCR-ABL融合蛋白。ELVN-001还具有针对耐药性最强的BCR-ABL1耐药突变体T315I和其他已知耐药突变体的活性。截至2025年4月28日的数据,在53例可评估的患者中,47%(25/53)在24周时达到主要分子学缓解(MMR),其中最后一线治疗时对酪氨酸激酶抑制剂(TKI)耐药的患者中有41%(14/34)在24周时处于MMR,曾接受asciminib或ponatinib治疗的患者中也有35%(12/34)在24周时处于MMR。所有达到或维持MMR的患者在数据截止时仍保持缓解。在该临床试验中所纳入的患者群体此前接受过大量治疗的情况下,ELVN-001与已获批的针对BCR-ABL融合蛋白的TKI在1期试验中所观察到的MMR相比,结果依然更好。安全性方面,ELVN-001在所有剂量水平上继续表现出良好的安全性和耐受性。
(2)Biomea Fusion公司公布BMF-500 1期临床试验初步数据
6月23日,Biomea Fusion公司公布了其共价FLT3抑制剂BMF-500治疗复发/难治性急性白血病的1期临床试验初步数据。BMF-500是一种新型、口服可利用的、高效、具选择性的FLT3共价小分子抑制剂。此前的研究表明,BMF-500潜在的脱靶风险较小,对活化的FLT3突变(包括FLT3-ITD和各种酪氨酸激酶结构域突变)具有皮摩尔级的亲和力。在携带FLT3-ITD突变的急性髓系白血病(AML)小鼠模型中,BMF-500使小鼠的肿瘤完全消退,且不需要持续暴露即可维持疗效。BMF-500在治疗FLT3突变的复发/难治性急性白血病患者中表现出令人鼓舞的初步疗效。在可评估的11例FLT3突变患者中,9人出现骨髓原始细胞下降,5人降幅超过50%。同时,在FLT3野生型患者中也观察到疾病控制效果。所有接受治疗的FLT3突变患者(n=18)的中位总生存期(mOS)为3.8个月(无CYP3A4抑制剂)和3.5个月(有CYP3A4抑制剂)。该数据优于历史对照数据,接受过gilteritinib和venetoclax治疗失败后的复发/难治性FLT3突变AML患者的历史mOS仅为2.1个月。BMF-500在各个剂量水平上总体耐受良好,未报告剂量限制性毒性、QT间期延长或因治疗相关不良事件导致的停药。
(3)Panavance Therapeutics公司公布misetionamide 1期临床试验中期数据
6月23日,Panavance Therapeutics公司公布了其小分子疗法misetionamide(GP-2250)联用吉西他滨治疗晚期不可切除或转移性胰腺导管腺癌患者1期临床试验的中期数据。Misetionamide具有独特的作用机制,可抑制两种致癌转录因子c-MYC和NFκB。此次公布的结果显示,与单独使用吉西他滨的历史结果相比,接受misetionamide联用吉西他滨治疗患者的确认的缓解率、疾病稳定率和无进展生存期更优,约42%的患者达到部分缓解或疾病稳定。此外,misetionamide联用吉西他滨的安全性良好,与单独使用吉西他滨的预期毒性相比,没有明显的新毒性或加剧的毒性。
(4)Astria Therapeutics公司公布navenibart 1b/2期临床试验数据
6月23日,Astria Therapeutics公司公布了其血浆激肽释放酶单抗抑制剂navenibart治疗遗传性血管性水肿的1b/2期临床试验长期开放标签试验的数据。此次公布的结果显示,患者每月的发作率大幅降低,平均降幅达92%,降幅中位数达97%。频率为每3个月给药一次组别患者的平均每月发作率降低了95%,而每6个月给药一次组别患者的平均降幅为86%。安全性方面,navenibart总体耐受性良好,无严重的治疗伴发不良事件,无停药现象。
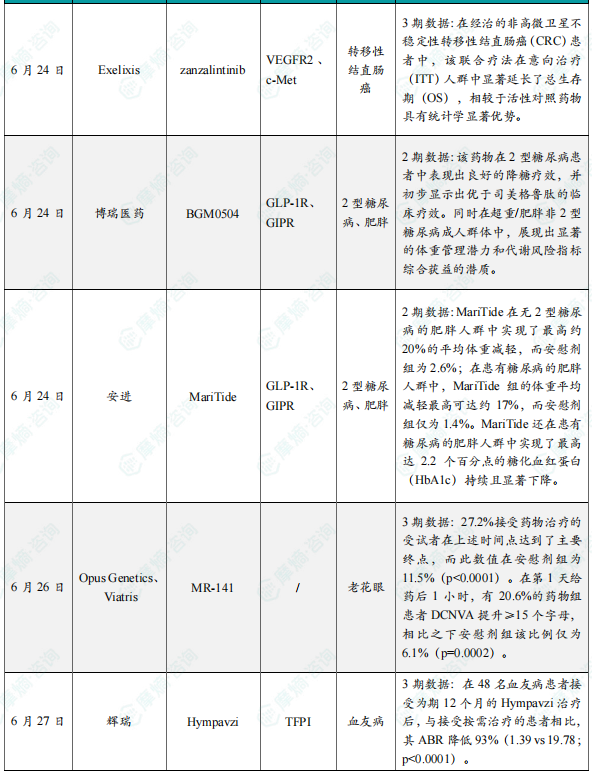
(5)Exelixis公司公布小分子抑制剂zanzalintinib 联合疗法3期临床试验积极结果
6月24日,Exelixis公司宣布,其小分子抑制剂zanzalintinib联合PD-L1抑制剂Tecentriq(atezolizumab)在STELLAR-303关键性3期试验中取得积极的顶线结果。在经治的非高微卫星不稳定性转移性结直肠癌(CRC)患者中,该联合疗法在意向治疗(ITT)人群中显著延长了总生存期(OS),相较于活性对照药物具有统计学显著优势。此次数据来自独立数据监查委员会对STELLAR-303研究双主要终点之一的最终分析结果。该研究将继续推进针对另一双主要终点——无肝转移患者(NLM)的总生存期分析。安全性方面,zanzalintinib联合atezolizumab的安全特征基本与以往观察一致,未发现新的安全性信号。这一结果进一步支持zanzalintinib作为此类转移性CRC患者的一种潜在治疗新选择。Zanzalintinib是一款新一代口服酪氨酸激酶抑制剂(TKI),靶向VEGF受体、MET以及参与肿瘤生长和免疫抑制的TAM激酶。值得一提的是,业内知名机构科睿唯安(Clarivate)在今年初发布的Drugs to Watch报告中,将zanzalintinib列为未来五年内有潜力成为重磅疗法的候选药物之一。
(6)博瑞医药公布BGM0504 2期临床试验积极数据
6月24日,博瑞医药在美国糖尿病协会(ADA)第85届科学会议上,公布了其自主研发的胰高血糖素样肽-1受体(GLP-1R)/葡萄糖依赖性促胰岛素多肽受体(GIPR)双靶点激动剂BGM0504的两项2期临床研究数据。两项独立的BGM0504 2期研究分别显示,该药物在2型糖尿病患者中表现出良好的降糖疗效,并初步显示出优于司美格鲁肽的临床疗效。同时在超重/肥胖非2型糖尿病成人群体中,展现出显著的体重管理潜力和代谢风险指标综合获益的潜质。BGM0504注射液是博瑞医药自主研发的GLP-1/GIP受体双重激动剂,可激动GIP和GLP-1下游通路,产生控制血糖、减重和治疗非酒精性脂肪性肝炎(NASH)等生物学效应,展现多种代谢疾病治疗潜力。该药物目前在中国开展针对体重管理和2型糖尿病的3期临床试验,并在美国完成US bridging临床研究。迄今已有超1000例患者接受治疗,显示出卓越疗效和良好安全性。
(7)安进公司公布在研药物MariTide 2期临床试验积极结果
6月24日,安进公司(Amgen)公布了其在研药物MariTide(maridebart cafraglutide)2期临床试验第一部分的完整结果。MariTide是一款潜在“first-in-class”的在研抗体多肽偶联药物,在靶向胃抑制肽受体(GIPR)的单克隆抗体的特定位点上偶联了两个GLP-1类似物,在激活GLP-1受体的同时抑制GIPR。它可通过皮下注射每月或更低频率给药。相关研究结果同时发表于《新英格兰医学杂志》。在2期研究中,MariTide在无2型糖尿病的肥胖人群中实现了最高约20%的平均体重减轻,而安慰剂组为2.6%;在患有糖尿病的肥胖人群中,MariTide组的体重平均减轻最高可达约17%,而安慰剂组仅为1.4%。在第52周时,体重减轻仍未出现平台期,显示出进一步减重的潜力。除了显著的减重效果外,MariTide还在患有糖尿病的肥胖人群中实现了最高达2.2个百分点的糖化血红蛋白(HbA1c)持续且显著下降。体重下降还伴随着一系列心血管代谢指标的改善,包括腰围、血压、高敏C反应蛋白(hs-CRP)及部分血脂参数的改善。
(8)Opus Genetics与Viatris公布老花眼眼药水MR-141关键3期试验积极结果
6月26日,Opus Genetics与Viatris宣布,其用于治疗老花眼的眼药水MR-141(phentolamine ophthalmic solution 0.75%)在第二项关键性3期临床试验VEGA-3中取得积极顶线结果。分析显示,该试验达到了主要终点和关键次要疗效终点,接受治疗的患者视力显著改善。值得关注的是,部分患者在用药后1小时内即出现明显视力提升,展现出该疗法的快速起效潜力。VEGA-3是一项多中心、随机、双盲、安慰剂对照的临床3期试验。共有545名受试者入组,按3:2比例随机接受每晚一次MR-141或安慰剂治疗。该试验的主要终点为与安慰剂相比,在第8天给药后12小时,受试者双眼远距矫正近视力(DCNVA)提高≥15个ETDRS字母(相当于≥3行),且双眼最佳矫正远视力(BCDVA)下降不足5个字母的比例。顶线分析结果显示,27.2%接受药物治疗的受试者在上述时间点达到了主要终点,而此数值在安慰剂组为11.5%(p<0.0001)。进一步分析显示,该药物在用药初期也能快速起效。在第1天给药后1小时,有20.6%的药物组患者DCNVA提升≥15个字母,相比之下安慰剂组该比例仅为6.1%(p=0.0002)。在第3天、第8天和第6周的多个时间点上,患者自我报告在清晨醒来后近视力满意度显著提升(p<0.0001),且近视力改善具有统计学显著性(p<0.0001)。而在第8天给药后12小时与第6周的数据对比中,未观察到快速耐受(tachyphylaxis)证据。
(9)辉瑞公布Hympavzi 3期临床积极结果,用于治疗血友病
6月27日,辉瑞(Pfizer)宣布其评估TFPI靶向抗体Hympavzi(marstacimab)用于体内存有抑制物的血友病A或血友病B成人和青少年患者的3期BASIS研究获得积极顶线结果。该研究达到了主要终点和关键的次要出血终点,结果显示每周一次皮下注射Hympavzi在改善关键出血结局方面优于按需治疗方案。辉瑞计划与监管机构讨论这些数据,目标是启动Hympavzi用于伴抑制物血友病患者的监管申请。分析显示,在伴抑制物的严重血友病A或B患者中,Hympavzi的预防性治疗可显著降低需要治疗的年化出血率(ABR),在统计学和临床上均具有显著意义。在48名血友病患者接受为期12个月的Hympavzi治疗后,与接受按需治疗的患者相比,其ABR降低93%(1.39 vs 19.78;p<0.0001)。Hympavzi在所有出血相关次要终点(自发性出血、关节出血、靶关节出血和总出血)上也显示出优势。该疗法总体耐受性良好,与过去的1/2期结果一致,未报告死亡或血栓栓塞事件。
(10)优时比小分子疗法芬氟拉明达3期试验主要终点
6月27日,优时比(UCB)宣布,其小分子疗法芬氟拉明(fenfluramine)在治疗CDKL5缺乏症(CDD)的3期临床试验中取得积极结果。CDKL5缺乏症是一种超罕见且严重的发育性和癫痫性脑病(DEE),其主要特征包括婴儿期起频繁发作的癫痫,以及严重的全球性神经发育迟缓,进而影响患者的认知功能、运动能力、皮层视觉功能以及睡眠质量。此次公布的研究为一项随机、双盲、安慰剂对照、固定剂量的多中心研究,共纳入87名年龄在1至35岁之间、确诊为CDD且癫痫未受控的患者,评估芬氟拉明作为辅助治疗的疗效与安全性。本研究的主要终点是比较芬氟拉明组与安慰剂组在药物滴定期及维持期内,与基线相比的可计数运动性癫痫发作频率(CMSF)中位百分比变化。研究结果显示,该试验已达到主要终点及大多数关键次要终点,且芬氟拉明具有良好的总体耐受性,其安全性与以往研究中观察到的结果一致。
同期事件:
1. 2025年第26周06.23-06.29国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2025年第26周06.23-06.29国内仿制药/生物类似物申报/审批数据分析
3.2025年第26周06.23-06.29国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2025.06.23-2025.06.29) },如需查看或下载完整版报告,可点击!
想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论