


































在很多人眼里,药物化学家似乎还停留在那种身穿白大褂、在充满刺鼻气味的实验室里摆弄烧瓶、一旦合成出新分子就大功告成的刻板印象中。然而,如果你读懂了现代药物研发的底层逻辑,就会发现这种认知已经落后了半个世纪。
今天的药物化学,早已不是单纯的“制造科学”,而是一门融合了生物学、药理学、计算机科学和物理化学的复杂艺术。它不再仅仅关注“我能合成什么”,而是更深刻地拷问“我应该合成什么,以及为什么”。
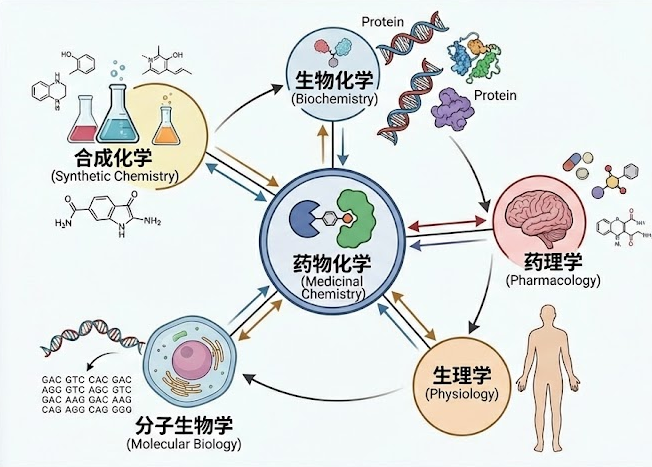
作为整个药物发现引擎的核心驱动力,药物化学家不仅是分子的建筑师,更是项目的战略家。他们必须在活性、选择性、代谢稳定性、生物利用度和安全性这几个常常相互冲突的维度中,寻找那个微妙而珍贵的平衡点。这注定是一场充满挑战的智慧博弈。
一、构效关系(SAR)
药物研发的终极目标,往往始于一个微小的化学结构变化。当我们面对癌症或脱发等生理挑战时,如何从亿万种化合物中找到那个“对”的分子?这就需要依赖构效关系(Structure-Activity Relationship, SAR)。
1. 分子的语言:结构决定命运
药物设计的核心在于处理分子的二元性:它必须对靶点有效(构效关系,SAR),同时也必须能够在生物系统中被递送至该靶点(构性关系,SPR)。
构效关系(SAR):
SAR研究的是化学结构的变化如何影响其生物活性。这通常被描述为“锁钥模型”或“诱导契合”关系,药物(钥匙)必须具备精确的电子和立体特征才能与蛋白靶点(锁)相互作用。然而,这种关系很少是线性的。引入一个甲基可能使效力提高一个数量级,也可能因为造成立体冲突而完全消除活性。这种微小的结构变化对生物学结果的巨大影响,是药物化学中最迷人也最令人沮丧的特征。
构性关系(SPR):
SPR则解决了药物的物理化学现实。一个对靶点具有皮摩尔级亲和力的分子,如果它在水中不溶解、无法穿透细胞膜,或者在几分钟内就被肝脏代谢殆尽,那么它作为药物的价值就为零。SPR涉及优化溶解度、渗透性(Papp)、亲脂性(log P)和拓扑极性表面积(TPSA)。药物化学家的挑战在于SAR和SPR往往是矛盾的;增强与疏水性靶点结合的亲脂性基团,往往会破坏分子的溶解度和代谢稳定性。
让我们通过一个具体的案例来理解这种精妙的微调。
假设我们有一个小分子药物,它的苯环部分需要嵌入靶蛋白的“结合口袋”中。
- 如果苯环朝向溶剂区(口袋外部): 无论你在苯环上挂什么装饰品(取代基),对活性的影响都微乎其微。
- 如果苯环深入口袋内部: 局势瞬间改变。此时,电子效应将主宰一切。
研究发现,在这个特定的口袋中,吸电子基团(如氯、三氟甲基)能显著增强活性,而供电子基团(如甲基、甲氧基)则会降低活性。
- 引入硝基(强吸电子),活性可能提升数千倍。
- 引入甲氧基(供电子),活性可能跌入谷底。
这就是SAR的威力——它告诉我们在哪里做加法,哪里做减法。
空间位阻的红线:
除了电子效应,大小也至关重要。结合口袋是有物理边界的。如果我们不断增大取代基的体积(如从甲基变成异丙基,再到叔丁基),起初可能会增加疏水作用力而提升活性;但一旦体积超过了“口袋”的红线,活性就会断崖式下跌。这就像试图把一只大象塞进冰箱,无论大象多么完美,尺寸不对就是不行。
二、手性的作用
生物系统本质上是手性的。蛋白质由L-氨基酸构成;DNA由D-糖构成。因此,药物靶点呈现出一种不对称的结合环境,对手性异构体(镜像分子)具有极高的识别敏感度。
手性的概念早在1893年由开尔文(Kelvin)定义:“如果任何几何图形或点集与其自身的镜像无法完全重叠,那么这个图形或点集就具有手性”。在药物发现中,这种几何属性绝非细枝末节——它往往是良药、安慰剂与毒药之间的区别。
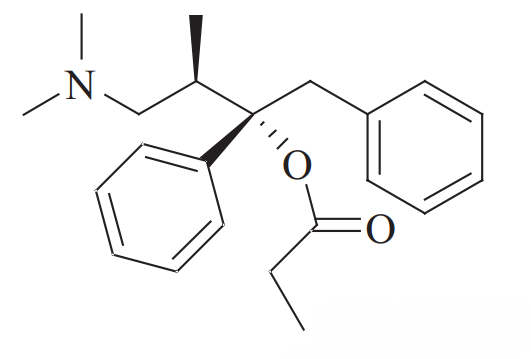
1. 丙氧芬悖论
关于手性识别最引人注目的历史案例之一涉及药物丙氧芬(Propoxyphene)。该分子具有两个手性中心,从而产生不同的立体异构体。
右丙氧芬(Dextropropoxyphene,商品名 Darvon): 其右旋异构体((2S, 3R)-型)是一种μ-阿片受体激动剂。它被广泛用作治疗轻中度疼痛的镇痛药。其三维形状与阿片受体的结合口袋互补,触发镇痛(及镇静)作用。
左丙氧芬(Levopropoxyphene, 商品名 Novrad): 其左旋异构体((2R, 3S)-型)对阿片受体的亲和力微乎其微,作为止痛药基本无效。然而,它却表现出强效的镇咳活性。
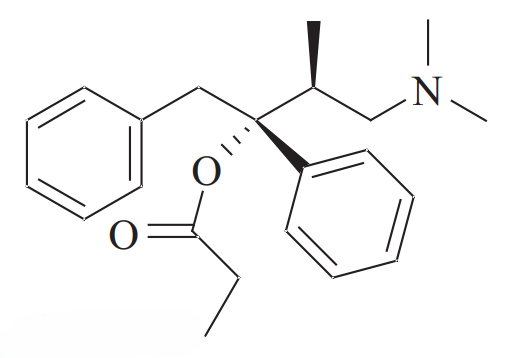
命名的讽刺艺术: 制造商礼来公司(Eli Lilly)在品牌命名中巧妙地利用了这种镜像关系。DARVON 倒过来拼写正是 NOVRAD 。这种命名法深刻地强调了立体化学的基本真理:对映异构体是具有截然不同生物宿命的独立化学实体。
2. 奥司他韦(Tamiflu)
绝对立体化学的重要性在抗流感药物 奥司他韦(Oseltamivir, 达菲)的设计中得到了生动的体现。奥司他韦是一种神经氨酸酶抑制剂,旨在模拟唾液酸裂解的过渡态。
靶点机制:流感病毒神经氨酸酶是病毒从感染细胞中释放新病毒颗粒所必需的表面酶。
结构设计:奥司他韦的活性代谢产物 GS-4071 包含一个庞大的疏水性戊基醚侧链和一个氨基。X射线晶体学显示,GS-4071 紧密结合在酶的活性位点内。
立体化学的决定性:乙酰胺基团和侧链的取向由环己烯环上的手性中心严格控制。如果翻转其中一个手性中心(例如翻转酰胺键),取代基将不再指向其指定的亚口袋,而是向外突出,与酶的内壁发生激烈的“立体碰撞”。这种碰撞会阻止抑制剂进入位点,使药物失效。奥司他韦的高效力直接归功于其特定的3D构型完美契合了神经氨酸酶的“负空间”。
三、定量构效关系(QSAR)
虽然定性的SAR(寻找诸如“氯比氢好”的趋势)很有用,但现代药物发现需要更高的精度。这导致了定量构效关系(QSAR)的发展——试图用数学方法将分子的物理性质与生物效力关联起来。
1. 从哈米特到汉施:数学化的飞跃
QSAR的根源在于物理有机化学。1937年,Louis Plack Hammett 提出了哈米特方程(Hammett Equation),将苯甲酸酯的水解速率与环取代基的电子性质(σ)相关联:
log(K/K0)= ρσ
这个方程量化了吸电子基团(正σ)或供电子基团(负σ)如何影响化学反应性。虽然这对化学来说是革命性的,但它无法解释生物系统,因为生物体比烧杯中水解的酯要复杂得多。
汉施方程(Hansch equation):突破来自波莫纳学院(Pomona College)的 Corwin Hansch。他意识到生物学增加了一个关键维度:转运。药物要起作用,必须穿过亲脂性的细胞膜。Hansch 将 Hammett 的电子参数与一个新术语结合起来:疏水性(亲脂性),用辛醇-水分配系数的对数(log P)来衡量。
经典的汉施方程通常采用以下形式:
log(1/C)=alogP+b(logP)2 +ρσ+δEs+d
ρ:反应常数
σ:只与取代基的电性有关的取代基常数。
δ:反应对位 阻效应的敏感性因子。
Es:位阻效应。
logP:底物的辛 醇/水分配系数;
a、b和d:通过实验数据的线性回归分析得出的常数;
C:代表测量时对应的底物浓度。
这个方程允许化学家预测未合成的分子的活性。如果模型显示活性与 log P 呈抛物线关系且最佳值为 3.5,化学家就会停止制造极性化合物,转而专注于亲脂性化合物。作为以数学方法量化药物及其靶点生物大分子相互作用的先驱之一,其被称为QSAR之父。
2. 托普利斯树:算法化的化学直觉
在高性能计算机普及之前,John Topliss(1970年代)设计了一种非数学的“决策树”,以便手工应用 Hansch 的原理。这种 托普利斯树 指导化学家在不运行回归分析的情况下进行结构优化。
决策流程(芳香取代示例):
① 起点:合成先导物的 4-氯(4-Cl)类似物。
② 评估:它比未取代的母体(H)更有效吗?
是(更有效):
这表明亲脂性增加(+π)和/或吸电子效应(+σ)是有益的。
下一步: 合成 3,4-二氯 类似物,以进一步增加亲脂性和吸电子能力。
否(较无效):
活性丧失。
检查: 是因为吸电子吗?尝试 4-OCH3(供电子)。
检查: 是因为立体位阻吗?尝试 4-CH3(较小的亲脂基团)。
这种启发式方法虽然比全QSAR简单,但它防止了在化学空间中的“随机游走”,确保每一个合成的分子都在测试关于蛋白需求的特定假设。
3. 现代展望:AI与深度学习的融合
进入21世纪20年代,QSAR已经从简单的线性回归进化为基于深度学习的复杂模型。图神经网络(GNNs)现在可以直接从分子图中学习特征,而无需人工设计的描述符。结合AlphaFold 3等工具,现在的QSAR不仅预测活性,还能预测蛋白-配体复合物的动态构象,将Hansch的静态方程升级为动态的生物物理模拟。
四、药效团
一个先导化合物很少是活性的整体;它是必需部分和非必需部分的组合。
- 药效团:IUPAC将其定义为确保与特定生物靶点进行最佳超分子相互作用并触发(或阻断)其生物反应所必需的立体和电子特征的集合。它是药物的“灵魂”——活性的最小核心骨架。
- 辅助基团:也就是“脚手架”,用于将药效团固定在适当的空间位置,或者调节物理化学性质(如溶解度、药代动力学),而不直接参与结合位点的关键相互作用。
1. 阿片类药物的解构:药效团的极简主义
阿片类镇痛药的历史提供了区分药效团与辅助基团的教科书级案例。
1. 吗啡(Morphine):
天然产物。具有复杂的五环刚性结构。强效。
2. 左啡诺(Levorphanol):
合成移除了醚桥(E环)和6-羟基。结果:保留了效力(甚至比吗啡强3-4倍)。结论:醚桥和6-OH不是关键药效团的一部分;它们甚至可能在空间上阻碍结合。
3. 苯并吗啡烷类(如美他佐辛 Metazocine):
进一步剥离C环。活性保留。
4. 哌替啶(Meperidine/Pethidine):
激进的简化。移除了B环和C环,只留下苯基哌啶核心。结果:虽然效力降低(约为吗啡的10-12%),但仍保留了镇痛活性并结合同一受体。
结论:阿片类药物的药效团并非复杂的吗啡骨架,而是一个叔胺基团与一个苯环通过碳链隔开的特定空间排列。这一发现使得设计纯合成阿片类药物(如芬太尼、哌替啶)成为可能,这些药物更易于制造且具有独特的药代动力学特征。
2.“反药效团”:hERG 规避与特非那定悲剧
药效团模型不仅用于寻找靶点,也用于规避反靶点。最臭名昭著的反靶点是 hERG 钾离子通道。阻断 hERG 会导致 QT 间期延长和致命的心律失常(尖端扭转型室速)。
- hERG 药效团特征:
大型、亲脂性且带有碱性胺的分子往往容易结合 hERG。
- 案例研究:特非那定(Terfenadine)
这种抗组胺药(商品名 Seldane)因阻断 hERG 导致心源性猝死而被撤市。它的继任者 非索非那定(Fexofenadine) 是其体内的羧酸代谢物。在结构中引入极性的羧酸盐(形成两性离子)破坏了 hERG 药效团(降低了亲脂性并改变了结合模式),在保留抗组胺活性的同时彻底消除了心脏毒性。这是利用结构修饰规避毒性药效团的经典案例。
五、筛选与设计策略
有了理论基础,我们如何获得第一个先导化合物?
1. 暴力美学 vs. 虚拟智慧
高通量筛选 (HTS):也就是“大海捞针”。利用自动化机器人,对几十万甚至上百万个化合物进行实体测试。这能发现全新的结构,但成本极其高昂。
虚拟筛选 (Virtual Screening):在超级计算机中,将数亿个分子(如Zinc数据库)与靶点模型进行“对接”。这大大节省了成本,但需要后续的实体实验来验证真伪。
2. 基于片段的药物设计
这是一种更精细的策略。我们不直接筛选大分子,而是筛选分子量很小(100-250)的片段。
- 这些片段结合力可能很弱,但如果我们将两个结合在相邻位点的片段通过化学键链接起来,其活性往往会呈指数级增长(协同效应)。
靶点导向动力学合成(Click Chemistry in situ):这是一个天才般的想法。让靶点蛋白自己“组装”它的抑制剂。如果两个反应基团在靶点口袋中靠得足够近,它们就会发生反应生成新分子。例如,利用胰岛素降解酶(IDE)作为模具,让叠氮化物和炔烃在酶的口袋里反应生成高活性的三唑类抑制剂。
六、生物电子等排原理
当一个先导化合物存在缺陷——代谢不稳定、毒性大或溶解度差——药物化学家就会求助于生物电子等排。这意味着用另一个基团替换现有功能基团,以保留相同的生物活性,但改变物理化学性质。
1. 经典与非经典等排体
经典等排体:具有相同价电子构型或原子数的基团。
- 例:用氟(F)替换氢(H)。氟在立体大小上与氢相似,但在电子性质上截然不同(高电负性),且代谢极其稳定(C-F键比C-H键强得多),常用于封闭代谢位点。
- 例:用 -NH2 或 -SH 替换 -OH。
非经典等排体:结构上看起来不同,但模拟原始基团的功能(如pKa、氢键模式、空间占位)。
2. 四唑的变形记:氯沙坦(Losartan)的故事
氯沙坦的开发是非经典生物电子等排的终极案例。
- 问题:早期的血管紧张素II受体拮抗剂含有羧酸基团(-COOH)。羧酸极性大(不利于膜渗透),且易被代谢(葡萄糖醛酸化)。
- 方案:化学家将 -COOH 替换为 四唑(Tetrazole) 环。
- 结果:四唑是一个平面的酸性杂环。它的 pKa(~4.9)与羧酸相似,因此在生理pH下以阴离子形式存在,维持了与受体的静电相互作用。然而,负电荷分散在四个氮原子上,使其具有更好的亲脂性和代谢稳定性,效力比酸高10倍。这一替换创造了第一个口服有效的沙坦类降压药,根据摩熵医药数据库显示,氯沙坦在多个国家批准上市。
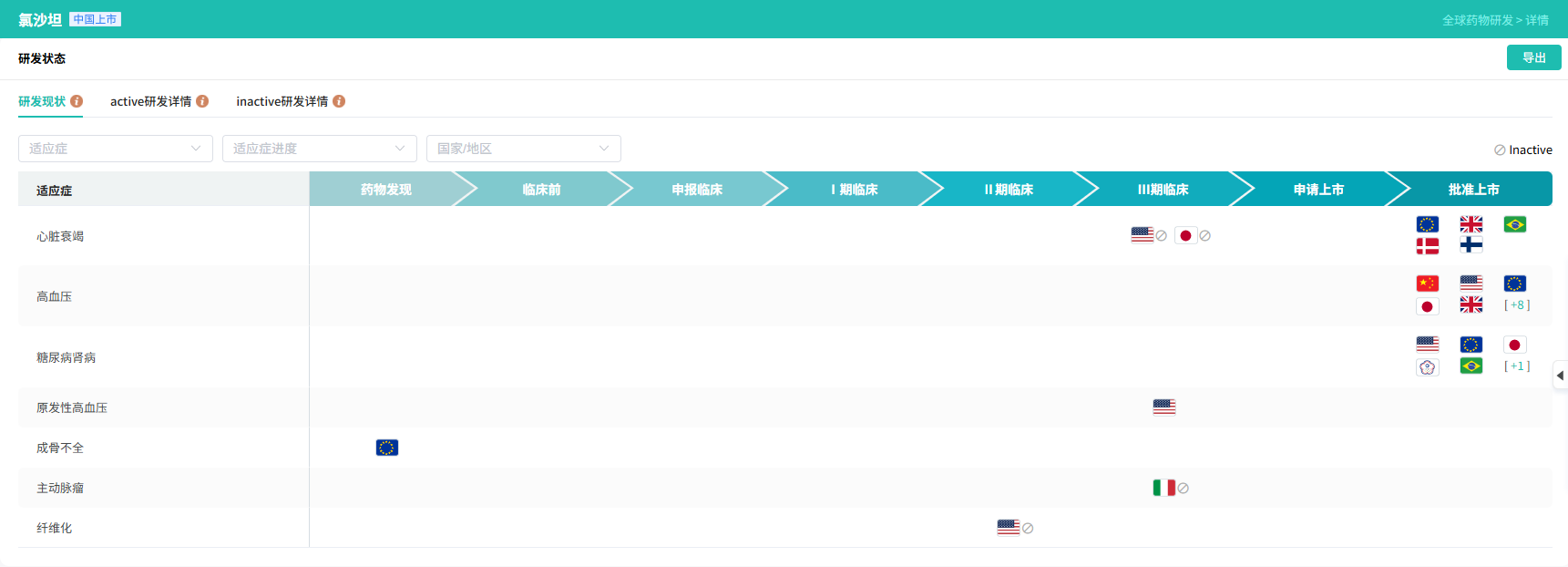
查数据,找摩熵!截图来源:摩熵医药数据库
七、通往成药之路—— 构效关系循环与类药性
1. 永无止境的循环
药物研发不是线性的,而是一个闭环:
设计 → 合成 → 测试 → 分析 → 再设计
每一轮循环,我们都希望离完美更近一步。保留提升活性的修饰,剔除降低活性的部分,直到获得理想的候选药物。
2. 利平斯基五规则
虽然化学空间浩瀚无垠(理论上可合成1060个分子),但能成为口服药物的分子往往集中在特定的“类药空间”。辉瑞的利平斯基( Christopher Lipinski )通过分析数千个药物,总结出了著名的类药五原则(Rule of 5, Ro5):
(1)分子量 < 500
(2)LogP(亲脂性) < 5
(3)氢键供体 < 5
(4)氢键受体 < 10
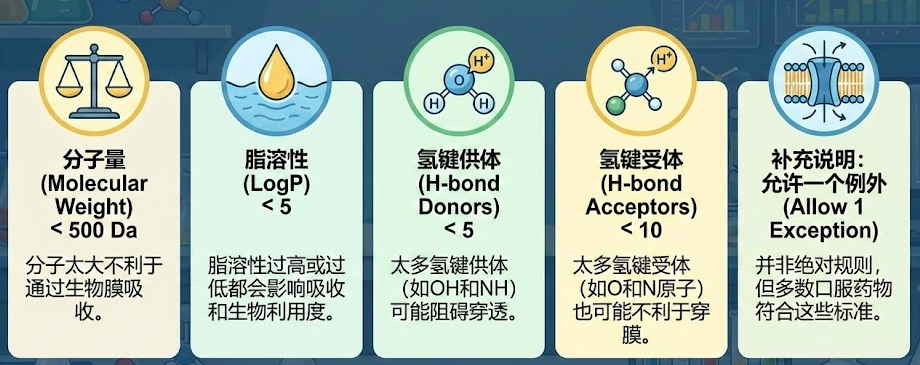
虽然这只是经验法则,并非铁律,但它时刻提醒我们:活性不是万能的,药物必须能被身体吸收和转运。
3. 超越五规则(bRo5)与 PROTACs
进入2020年代,教条正在发生转变。PROTACs(蛋白降解靶向嵌合体)和“分子胶”等新模态故意违反五规则。这些分子的分子量往往超过 800 Da,目的是降解蛋白而非仅仅抑制它们。
- 变色龙效应:
尽管体积巨大,一些 PROTACs 实现了口服生物利用度。它们通过形成分子内氢键,在穿过细胞膜时“折叠”起来隐藏极性基团(像变色龙一样伪装),然后在细胞内展开。
- 新范式:
药物化学正在从“遵守五规则”转向“五规则意识”,承认规则是指南,而非物理定律。对于难成药靶点,打破规则往往是必须的。
结语
现实中几乎不存在完美的分子,每一个上市药物都是无数次妥协与优化后的幸存者。正如我们文中提到的,药物化学家利用SAR、QSAR和生物电子等排等工具,在原子尺度上精雕细琢,解决了“结合靶点”的问题。
但请记住药物研发领域那句残酷的格言:“活性(Activity)是药物的入场券,性质(Property)才是药物的各种死法。”
哪怕一个分子对靶点有皮摩尔级的亲和力,如果它无法被肠道吸收(A),无法分布到靶器官(D),瞬间被代谢殆尽(M),或者排泄半衰期过短(E),它依然无法成为药物。这也是为什么数以万计的高活性分子最终倒在了临床前阶段的“死亡之谷”。
【往期精彩】
1. 药物研发“硬核攻略”:ADME过程、体外模型构建及体内PK参数全解析
2. 干货 | 药物筛选五次技术迭代:从表型到AI,虚拟筛选战术图谱大揭秘
3. 揭秘药物研发新纪元:物理载体、软件逻辑、AI架构与数据挑战全解析
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论