


































1.4.1 本周全球TOP10创新药研发进展

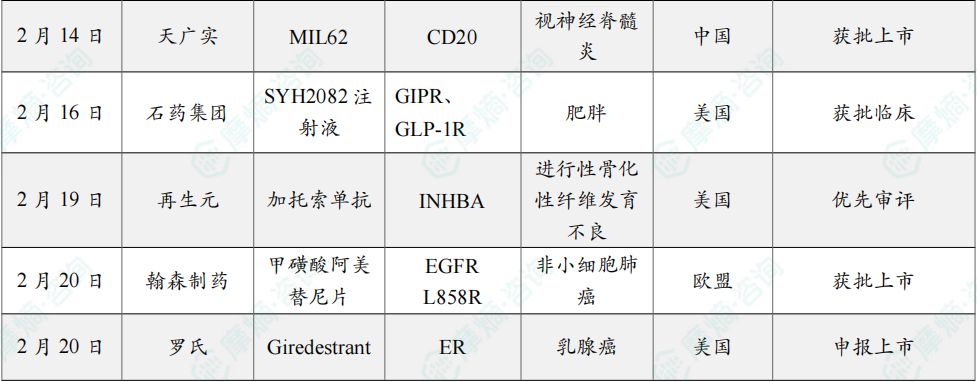
(1)诺诚健华分子胶1类新药获批临床
2月9日,诺诚健华宣布,公司自主研发的VAV1分子胶降解剂 ICP-538 获中国国家药品监督管理局(NMPA)药品审评中心(CDE)批准开展临床研究,拟开发治疗多发性硬化(MS)。
ICP-538 是新型口服高效、高选择性靶向VAV1的特异性分子胶降解剂,拟用于开发治疗多种难治的自身免疫性疾病,比如炎症性肠病(IBD)、系统性红斑狼疮(SLE)和MS。ICP-538 通过高效选择性介导CRBN E3泛素连接酶与VAV1蛋白形成三元复合物,剂量依赖性地诱导VAV1蛋白快速高效降解。VAV1是T细胞和B细胞受体下游的关键蛋白,降解VAV1可以有效抑制T细胞增殖、分化、激活及细胞因子释放,并抑制B细胞激活与细胞因子释放,从而发挥抗炎及免疫调节作用,缓解自身免疫和炎症的病理进程。
临床前研究表明,ICP-538 深度降解VAV1,导致与免疫介导病症相关的细胞因子显著减少,而对其他蛋白没有可检测到的影响。
(2)和誉医药FGFR4抑制剂获FDA快速通道资格,治疗肝癌
2月10日,和誉医药宣布,美国FDA已授予该公司自主研发的高选择性小分子FGFR4抑制剂 依帕戈替尼(irpagratinib/ABSK-011)快速通道资格,用于既往接受过免疫检查点抑制剂(ICI)和多靶点激酶抑制剂(mTKI)治疗,且存在FGF19过表达的肝细胞癌(HCC)患者。
依帕戈替尼 是和誉医药自主开发的一款高选择性FGFR4抑制剂,用于治疗FGF19过表达的晚期HCC患者。此次被FDA授予FTD,主要基于公司在2024年欧洲肿瘤内科学会(ESMO)年会上公布的1期临床研究积极结果。数据显示,在既往接受过ICI和mTKI治疗失败且FGF19过表达的晚期HCC患者中,依帕戈替尼 单药治疗展现出显著疗效及良好的安全性和耐受性,客观缓解率(ORR)达到46.7%,中位无进展生存期(mPFS)为5.5个月。
此外,和誉医药在2025年欧洲肿瘤内科学会胃肠道肿瘤大会(ESMO-GI)上公布了 依帕戈替尼 联合抗PD-L1抗体 阿替利珠单抗 治疗HCC的最新研究结果。在初治及既往接受过ICI治疗的FGF19过表达HCC患者中,该联合治疗方案的ORR均超过50%,mPFS超过7个月,且未观察到新的安全性信号,显示出冲击肝细胞癌一线治疗方案的潜力。
(3)礼来的米吉珠单抗在中国获批双适应症
2月11日,礼来公司(Eli Lilly and Company)宣布,米吉珠单抗 两个剂型获得中国国家药品监督管理局(NMPA)批准,用于治疗成人中重度活动性克罗恩病(CD)及成人中重度活动性溃疡性结肠炎(UC)。
米吉珠单抗 是一种特异性靶向白介素-23(IL-23) p19亚基的IgG4型单克隆抗体,可选择性抑制IL-23通路,调节由其驱动的免疫炎症反应。
该药于2023年获批用于治疗中重度活动性UC,目前已在包括美国、欧盟和日本在内的多个国家和地区获批用于治疗中重度活动性UC和CD。
(4)首款JAK抑制剂凝胶申报上市,来自普祺医药
2月11日,CDE网站显示,普祺医药的 普美昔替尼(PG-011)凝胶申报上市,用于治疗特应性皮炎。该药物有望成为特应性皮炎领域首款JAK抑制剂凝胶,并且也是首款JAK抑制剂凝胶。
普美昔替尼 是普祺医药自主研发的一款JAK1/JAK2抑制剂,可高效抑制IL-4、IFN-γ、IL-6、IL-12、IL-1β及TGF-β1等多种炎症因子,与同类药物相比覆盖炎症因子范围更广。通过抑制更多与炎症相关的因子,普美昔替尼 能够有效地从源头阻断炎症信号传导。
普祺医药就 普美昔替尼 开发了凝胶和鼻喷雾剂两种剂型,其中 普美昔替尼凝胶 已在成人及青少年特应性皮炎人群中完成一项III期临床试验(PG-011-AD-301),普美昔替尼鼻喷雾剂 正在开展治疗季节性过敏性鼻炎的III期临床试验,有望成为该领域首款JAK抑制剂鼻喷雾剂。
普祺医药拟于2026年第四季度提交该鼻喷雾剂的上市申请,济川药业拥有其在中国市场的10年独家商业化权益。
(5)三生国健自主研发的安沐奇塔单抗注射液获批上市
2月13日,三生国健宣布,公司自主研发的1类生物创新药,抗IL-17A人源化单克隆抗体 安沐奇塔单抗注射液(商品名:益赛拓®)上市申请已获国家药品监督管理局批准,用于适合系统治疗或光疗的中度至重度斑块状银屑病成人患者。
安沐奇塔单抗提供两种灵活的用药选择,维持期患者可实现每4周或每8周一次的给药频率。安沐奇塔单抗维持期每8周一次的给药方案仍可维持疗效强效应答,相较于现有同靶点抑制剂的治疗方案,给药间隔更长。在不影响疗效和安全性的基础上,安沐奇塔单抗每8周一次的简化给药方案有望降低患者年度用药频率,有效降低长期治疗带来的时间成本与心理压力。
(6)天广实三代CD20单抗MIL62获批上市
2月14日,天广实宣布其自主研发的创新型第三代CD20抗体药物 MIL62(商品名:倍捷欣,通用名:奥妥珠单抗β注射液)获批上市,用于治疗视神经脊髓炎谱系疾病(NMOSD)。
MIL62 是天广实自主研发的一种创新型的II型抗CD20重组人源化单克隆抗体,采用了独特的岩藻糖全敲除技术增强抗体ADCC,是中国首款国产第三代CD20抗体。
在临床前体外及体内研究中,与第一代抗CD20抗体 利妥昔单抗 相比,MIL62 表现出更强的ADCC活性和清除体内异常激活B细胞的能力。
(7)石药集团每月一次减重多肽新药在美国获批临床
2月16日,石药集团宣布,该公司开发的GLP-1/GIP受体双偏向性激动多肽长效注射液(SYH2082注射液)已获得美国FDA批准,可在美国开展临床试验。SYH2082 有望成为一款长效GLP‑1/GIP受体双重偏向性激动剂,每月给药一次。依托长效制剂技术平台,SYH2082 实现月度给药,提高患者依从性与用药便捷性。
根据石药集团新闻稿介绍,SYH2082 可选择性激活cAMP通路,降低β-arrestin募集,从而减少受体内吞及脱敏,提升药效并延长作用持续时间。同时,SYH2082 结合长半衰期修饰平台技术和长效制剂平台技术,旨在实现给药间隔内的持续减重。
在临床前研究中,SYH2082 在长效减重及维持方面较同类上市产品表现出更优的疗效,且支持每月一次的用药方案。在毒理学研究中,SYH2082 的耐受性良好,未观察到显著不良反应。
本次获批的临床适应症为肥胖或超重合并至少一种体重相关合并症人群的体重管理。此外,SYH2082 亦具备改善成人2型糖尿病(T2DM)患者的血糖控制的潜力,带来额外临床获益。
(8)再生元Actvin A单抗获FDA优先审评,治疗进行性骨化性纤维发育不良
2月19日,再生元宣布 加托索单抗(garetosmab)的生物制品许可申请(BLA)获FDA优先审评,用于治疗成人进行性骨化性纤维发育不良(FOP),PDUFA日期为2026年8月。若获批,garetosmab 将成为全球首款能减少成人FOP患者新异位骨(heterotopic ossification,HO)病变数量和体积的治疗药物。
FOP是由ACVR1基因突变驱动的超罕见遗传性疾病,全球确诊仅约900例。该疾病的核心特征为肌肉、肌腱等结缔组织逐步被异常骨组织取代,累及颌骨、脊柱等关键部位时,会导致患者进食、呼吸、行动障碍,多数患者30岁前便需依靠轮椅,目前临床上尚无有效治疗手段。
值得关注的是,garetosmab 此前已获FDA快速通道认定和孤儿药认定,欧盟也授予其孤儿药资格,再生元还计划在全球多国提交该药的监管申请。此外,针对儿童和青少年FOP患者的III期OPTIMA 2试验也将于2026年启动。
(9)翰森制药肺癌新药阿美替尼于欧盟获批上市
2月20日,翰森制药宣布,创新药 甲磺酸阿美替尼片 单药治疗已正式获得欧盟委员会(EC)批准在欧盟上市,用于:(i)具有表皮生长因子受体(EGFR)外显子19缺失或外显子21(L858R)置换突变的晚期非小细胞肺癌(NSCLC)成人患者的一线治疗;(ii)晚期EGFR T790M突变阳性NSCLC成人患者的治疗。本次获批是在欧洲药品管理局(EMA)人用药品委员会(CHMP)发布积极意见之后正式作出的。
作为翰森制药自主研发的三代EGFR-TKI,甲磺酸阿美替尼 具有良好的脂溶性和稳定性,能更好地透过血脑屏障,且不良反应发生率低。
该产品自2020年上市以来,持续深耕NSCLC治疗领域,不断拓展适应症边界。截至目前,阿美替尼 已有五项适应症获中国NMPA批准上市,治疗范围全面覆盖:EGFR突变NSCLC患者的术后辅助治疗、不可切除局部晚期NSCLC放化疗后的维持治疗、靶化联合治疗局晚或转移性NSCLC一线治疗,以及单药用于晚期NSCLC的一线治疗和二线治疗。
(10)罗氏口服SERD药物在美国申报上市
2月20日,罗氏宣布 Giredestrant 的上市申请获得FDA受理,用于联合 依维莫司 治疗既往接受过内分泌疗法治疗且携带ESR1突变的ER+/HER2-局部晚期或转移性乳腺癌患者。
PDUFA日期为2026年12月18日。如果获批,该组合疗法将成为乳腺癌患者在CDK4/6抑制剂经治后可用的首个SERD全口服方案。
Giredestrant 是罗氏自主研发的一款下一代口服选择性雌激素受体降解剂(SERD)和完全拮抗剂,旨在阻止雌激素与雌激素受体(ER)结合,诱导其降解,从而阻止或减缓癌细胞的生长。
1.4.2 本周全球TOP10积极/失败临床结果
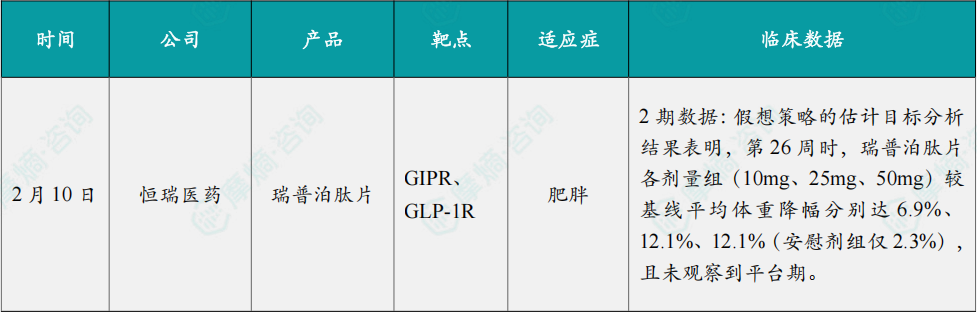
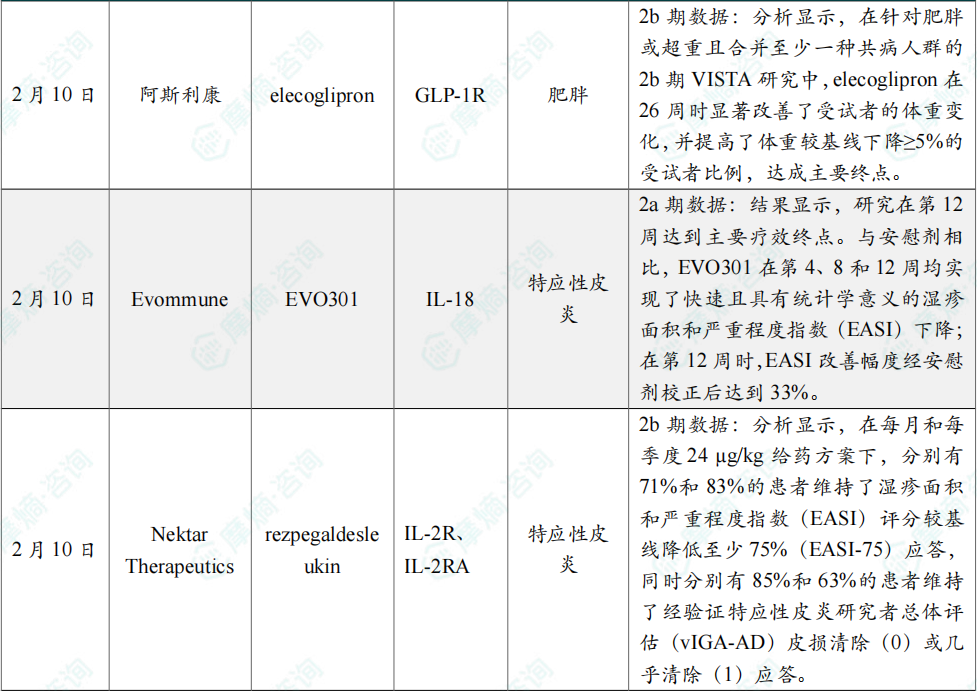


(1)恒瑞医药口服GLP-1R/GIPR激动剂II期研究成功
2月10日,恒瑞医药与Kailera Therapeutics共同宣布,每日1次口服 瑞普泊肽片 在中国166名肥胖成人中开展的II期临床试验(HRS9531-T-201)取得积极顶线数据。基于令人鼓舞的临床数据,恒瑞医药将在中国推进 瑞普泊肽片 进入III期试验,合作方Kailera计划于2026年启动全球II期试验。
HRS9531-T-201研究是一项由恒瑞医药在中国开展的多中心、随机、双盲、安慰剂对照II期临床试验(n=166),旨在评估 瑞普泊肽片(10/25/50mg,每日1次)对比安慰剂在不伴有2型糖尿病的肥胖(≥28kg/m2)成人受试者中的疗效和安全性。假想策略的估计目标分析结果表明,第26周时,瑞普泊肽片 各剂量组(10mg、25mg、50mg)较基线平均体重降幅分别达6.9%、12.1%、12.1%(安慰剂组仅2.3%),且未观察到平台期。疗法策略的估计目标分析结果表明,第26周时,瑞普泊肽片 各剂量组(10mg、25mg、50mg)较基线平均体重降幅分别为6.7%、11.9%、11.4%(安慰剂组仅2.1%),同样未观察到平台期。此外,第26周时,25mg剂量组达到减重幅度≥10%和≥15%的受试者比例分别为59.1%和38.6%;50mg剂量组达到减重幅度≥10%和≥15%的受试者比例分别为52.5%和37.5%。
(2)阿斯利康口服GLP-1减肥药2b期临床达主要终点
2月10日,阿斯利康(AstraZeneca)在公布的季报中披露,其口服GLP-1候选药物 elecoglipron(AZD5004)在两项分别用于治疗肥胖/超重人群及2型糖尿病患者(T2D)的2b期临床试验中均达成主要终点。公司表示,该试验结果支持 elecoglipron 启动3期临床试验。
分析显示,在针对肥胖或超重且合并至少一种共病人群的2b期VISTA研究中,elecoglipron 在26周时显著改善了受试者的体重变化,并提高了体重较基线下降≥5%的受试者比例,达成主要终点。在另一项面向2型糖尿病患者的2b期SOLSTICE研究中,该药物同样在26周时实现了糖化血红蛋白(HbA1c)较基线下降这一主要终点。2023年11月,阿斯利康与诚益生物达成独家协议,以潜在18.25亿美元的总额获得其开发权益,用于治疗包括肥胖症、T2D和其他合并症在内的适应症。
(3)Evommune特应性皮炎IL-18新型疗法2a期数据积极
2月10日,Evommune宣布,其针对中重度特应性皮炎(AD)成人患者的2a期临床试验取得积极顶线结果。该研究为一项随机、双盲、安慰剂对照试验,评估候选药物 EVO301 的安全性和有效性。
EVO301 是一款长效融合蛋白,由白细胞介素-18(IL-18)结合蛋白与抗血清白蛋白Fab相关结构域组成。该疗法旨在中和异常升高的IL-18活性,并在组织分布效率、结合亲和力和特异性方面,相较既往针对IL-18通路的拮抗或抑制策略展现出潜在优势。
本次试验共入组70例患者,采用静脉给药方案,在第1天和第28天分别给予5mg/kg剂量,随访周期为12周。结果显示,研究在第12周达到主要疗效终点。与安慰剂相比,EVO301 在第4、8和12周均实现了快速且具有统计学意义的湿疹面积和严重程度指数(EASI)下降;在第12周时,EASI改善幅度经安慰剂校正后达到33%。此外,23%的 EVO301 治疗患者在第12周实现了研究者总体评估(IGA)皮损清除(0)或几乎清除(1)的治疗目标。
(4)Nektar Therapeutics公布特应性皮炎药物2b期临床积极数据
2月10日,Nektar Therapeutics宣布,其用于治疗中重度特应性皮炎(AD)患者的创新调节性T细胞(Treg)靶向生物制品 rezpegaldesleukin,在REZOLVE-AD研究中为期36周的盲法维持治疗阶段取得积极结果。
REZOLVE-AD是一项全球性2b期研究,共入组393例中重度特应性皮炎患者。患者按3:3:3:2的比例随机分配,在16周诱导期内接受三种不同剂量的 rezpegaldesleukin 或安慰剂皮下注射治疗。完成16周诱导期后,接受 rezpegaldesleukin 治疗且达成EASI-50的患者,再次按1:1比例随机分配,在随后的36周盲法维持期内,以与诱导期相同剂量按每4周一次(Q4W)或每12周一次(Q12W)给药,直至第52周。
分析显示,在每月和每季度24µg/kg给药方案下,分别有71%和83%的患者维持了湿疹面积和严重程度指数(EASI)评分较基线降低至少75%(EASI-75)应答,同时分别有85%和63%的患者维持了经验证特应性皮炎研究者总体评估(vIGA-AD)皮损清除(0)或几乎清除(1)应答。
(5)诺华IgA肾病新药III期研究最终分析结果积极
2月13日,诺华宣布 阿曲生坦(英文商品名:Vanrafia,中文商品名:诺锐达)治疗IgA肾病的III期ALIGN研究在最终分析中达到了关键次要终点,证明其可以延缓IgA肾病患者的肾功能下降速度。基于此,诺华将在2026年向FDA申请将 阿曲生坦 用于治疗IgA肾病的加速批准转换为常规批准。
阿曲生坦 是诺华收购Chinook Therapeutics获得的一款高选择性内皮素A受体(ETA)拮抗剂。该药物最初由艾伯维发现,Chinook Therapeutics在2020年1月获得了其全球独家权益。2025年4月,阿曲生坦基于ALIGN研究的中期分析结果获得FDA加速批准上市,并在同年8月获得中国国家药监局加速批准上市。
中期分析结果显示,经 阿曲生坦 治疗后,IgA肾病患者最早在第6周即可观察到尿总蛋白肌酐比值(UPCR)降低。治疗第36周时,阿曲生坦 组患者的24h UPCR较安慰剂组降低了36.1%(P<0.0001)。
在此次最终分析中,治疗第4-136周,两组之间的估计肾小球滤过率(eGFR)较基线变化值的差异为2.39mL/min/1.73m2(双侧P=0.057)。此外,在研究结束时(即第132周),阿曲生坦 组患者的eGFR较基线变化值比安慰剂组增加了2.59mL/min/1.73m2(名义双侧P=0.039)。在预先指定的探索组(额外接受SGLT2抑制剂治疗)中,亦观察到临床意义显著的结果。
(6)赛诺菲公布TL1A单抗IIb期研究长期数据
2月17日,赛诺菲公布了 duvakitug 治疗溃疡性结肠炎(UC)和克罗恩病(CD)的IIb期RELIEF UCCD研究的长期分析结果。
Duvakitug 是梯瓦公司研发的一款靶向肿瘤坏死因子(TNF)样配体1A(TL1A)的人源化IgG1-λ2单克隆抗体。2023年10月,赛诺菲与梯瓦达成协议,获得该药物的合作开发与商业化权益。
本次更新的44周维持治疗研究数据显示,在UC队列中,duvakitug(450mg)组和 duvakitug(900mg)组分别有47%和58%的患者实现临床缓解;在CD队列中,duvakitug(450mg)组和 duvakitug(900mg)组分别有41%和55%的患者实现内镜缓解。安全性方面,duvakitug 组最常见的不良事件(发生率≥5%)为上呼吸道感染、鼻咽炎、CD和高血压,并且与诱导治疗研究数据一致。
(7)罗氏单抗疗法达肾病3期试验主要终点
2月17日,罗氏(Roche)宣布,其在成人原发性膜性肾病患者中开展的3期MAJESTY研究已达到主要终点。
研究结果显示,与活性对照相比,接受Gazyva/Gazyvaro(obinutuzumab)治疗的患者在两年(104周)时实现完全缓解的人数显著更多,差异具有统计学意义且具临床意义。安全性方面,研究结果与该药物既往已建立的安全性特征一致,未发现新的安全性信号。关键次要终点分析同样显示积极结果,包括在第104周总缓解率(完全缓解或部分缓解)以及第76周完全缓解率方面,obinutuzumab 均较活性对照展现出具有统计学意义和临床意义的改善。
罗氏预计将该试验结果与美国和欧盟监管机构分享。根据新闻稿,基于本次研究结果,该疗法有望成为首个获批用于治疗原发性膜性肾病的药物。
(8)辉瑞小分子结直肠癌药物3期结果积极
2月17日,辉瑞(Pfizer)宣布,其关键性3期BREAKWATER研究中队列3取得积极顶线结果。该队列评估了 Braftovi(encorafenib)与西妥昔单抗(商品名Erbitux)和mFOLFOX6治疗方案(包括氟尿嘧啶、亚叶酸钙和奥沙利铂)构成的组合疗法,用于既往未经治疗、携带BRAF V600E突变的转移性结直肠癌(mCRC)患者的疗效。
盲态独立中心评估(BICR)确认结果显示,与FOLFIRI治疗方案(联合或不联合贝伐珠单抗)相比,Braftovi 联合方案在关键次要终点无进展生存期(PFS)方面实现了具有统计学意义且具临床意义的改善。同时,在描述性次要终点总生存期(OS)方面,该联合方案亦展现出具有临床意义的延长趋势。此前公布的试验结果显示,该队列的主要终点——基于BICR评估的客观缓解率(ORR)同样取得积极结果。
(9)礼来替尔泊肽联合依奇珠单抗III期研究成功
2月18日,礼来宣布具有里程碑意义的TOGETHER-PsO开放标签IIIb期研究取得积极结果。该研究评估了依奇珠单抗 联合 替尔泊肽,对比 依奇珠单抗 单药治疗中重度斑块状银屑病合并肥胖或超重(且至少伴有一种体重相关合并症)成人患者中的疗效。
在这项首创性TOGETHER-PsO研究中,27.1%的 依奇珠单抗 联合 替尔泊肽 治疗患者实现了完全皮肤清除(银屑病面积和严重程度指数PASI 100)且体重下降至少10%,而 依奇珠单抗 单药治疗组仅为5.8%,达到主要终点(p<0.001)。在一项关键次要终点中,联合治疗组实现PASI 100的患者比例较 依奇珠单抗 单药治疗提高40%(分别为40.6%和29.0%,p<0.05),证实通过替尔泊肽治疗肥胖或超重可减轻银屑病疾病负担。
(10)默沙东长效单抗3期结果积极,有望扩展适应症
2月20日,默沙东(MSD)公布其3期SMART研究(MK-1654-007)的最新结果。该研究在两个RSV流行季中开展,旨在评估 Enflonsia(clesrovimab)这一长效预防性单克隆抗体在易发展为重症呼吸道合胞病毒(RSV)感染的儿童人群中的保护作用。
最新数据显示,在年龄不足2岁且在第二个RSV流行季仍处于重症风险中的儿童中,于第二个流行季开始时接受 Enflonsia 治疗后,其整体安全性表现良好。该结果与第一流行季接受该疗法的婴儿人群总体一致。
基于上述研究结果,默沙东计划将第二个RSV流行季的数据提交美国FDA及全球其它监管机构,用于支持该疗法在高风险儿童中拓展至第二个RSV流行季预防适应症的评估。
同期事件:
1. 2026年第7-8周02.09-02.22国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2026年第7-8周02.09-02.22国内仿制药/生物类似物申报/审批数据分析
3. 2026年第7-8周02.09-02.22国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2026.02.09-2026.02.22) },如需查看或下载完整版报告,可点击!
扩展阅读:
1. 2026年第6周02.02-02.08全球创新药研发概览
2. 2026年第5周01.26-02.01全球创新药研发概览
3. 2026年第4周01.19-01.25全球创新药研发概览
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论