


































1.4.1 本周全球TOP10创新药研发进展
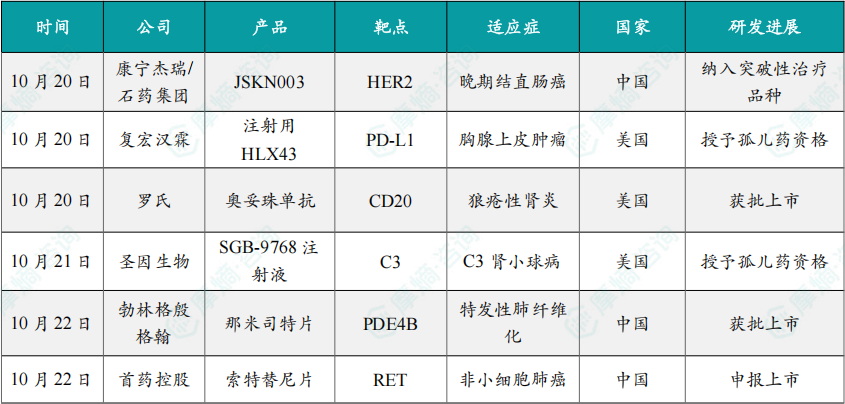
(1)康宁杰瑞/石药集团HER2双抗ADC新药JSKN003被纳入突破性治疗品种
10月20日,康宁杰瑞与石药集团共同宣布,HER2双特异性抗体偶联药物(ADC)JSKN003 再次获得中国国家药品监督管理局药品审评中心(CDE)突破性疗法认定,适应症为单药用于治疗既往经奥沙利铂、氟尿嘧啶和伊立替康治疗失败的HER2阳性晚期结直肠癌。
此前,JSKN003针对不限HER2表达水平的铂耐药卵巢癌适应症已获得CDE突破性疗法认定、美国FDA临床研究许可,胃癌及胃食管结合部癌适应症已获得FDA孤儿药资格。
目前,JSKN003正在中国开展用于治疗乳腺癌、卵巢癌、胃癌等实体瘤的多项2期及3期临床研究,JSKN003再次获授予突破性治疗认定,将进一步加快该产品的研发和审评速度,以期早日惠及更多患者。
2024年9月,康宁杰瑞与石药集团全资子公司津曼特生物达成授权合作,津曼特生物获得在中国内地(不包括香港、澳门及台湾地区)开发、销售、许诺销售及商业化 JSKN003 用于治疗肿瘤相关适应症的独家许可及再许可权。
(2)复宏汉霖PD-L1 ADC HLX43获美国FDA孤儿药资格认定
10月20日,复宏汉霖宣布,公司创新型程序性死亡-配体1(PD-L1)抗体偶联药物(ADC)注射用HLX43已获得美国食品药品监督管理局(FDA)授予的孤儿药资格认定(Orphan Drug Designation, ODD),用于胸腺上皮肿瘤(Thymic Epithelial Tumors,TETs)的治疗。
HLX43 是全球首个布局胸腺上皮肿瘤的PD-L1 ADC,其I期临床研究在胸腺癌(Thymic Carcinoma)等实体瘤中展现出“高效、低毒”的治疗潜力(2025 ASCO:75%的胸腺癌患者达到部分缓解,ORR=75%)。基于此,公司高效推进HLX43在中、美、日、澳等地的国际多中心临床研究,其用于TC治疗已获得中国、美国、日本等地监管机构的临床试验许可。
(3)罗氏的奥妥珠单抗获批治疗狼疮性肾炎
10月20日,罗氏宣布奥妥珠单抗(商品名:Gazyva/Gazyvaro)获FDA批准新适应症,用于治疗正在接受标准治疗的活动性狼疮性肾炎(LN)成人患者。在第一年接受四次初始剂量治疗后,患者的治疗频率可更改为一年两次。
该药物是首个获得FDA批准用于治疗狼疮性肾炎的CD20靶向疗法。
FDA此次批准主要是基于II期NOBILITY研究和III期REGENCY研究的积极结果。REGENCY研究显示,奥妥珠单抗联合标准治疗(霉酚酸酯和糖皮质激素)组有46.4%的患者在第76周时实现了完全肾脏缓解(CRR),而标准治疗组这一比例为33.1%(调整后差异为13.4%,95% CI:2.0%-24.8%;P=0.0232)。与此同时,奥妥珠单抗组的补体水平相较于标准治疗组也实现了具有临床意义的改善,并且抗dsDNA抗体、疾病活动度和炎症标志物水平也有所降低。
奥妥珠单抗于2013年在美获批上市,属于第三代Fc段经修饰的II型人源化CD20单抗。目前该药物已获批用于治疗3类血液瘤,包括慢性淋巴细胞白血病、滤泡性淋巴瘤、小淋巴细胞性淋巴瘤。
(4)圣因生物靶向C3 siRNA药物SGB-9768注射液获FDA孤儿药资格
10月21日,圣因生物(SanegeneBio)宣布,其自主研发的靶向补体C3的siRNA药物 SGB-9768注射液 近日获得美国FDA授予孤儿药资格,用于治疗C3肾小球病(C3 glomerulopathy,C3G)。
作为一种新型RNAi疗法,SGB-9768 凭借每年仅需两次的皮下给药方案和良好的安全性,有望显著改善C3G患者的生活。C3G是一种罕见的肾脏疾病,在全球范围内发病率为每年1~2例/100万人,多发于青壮年。约50%的患者在10年内进展至终末期肾病,需接受透析或肾移植手术;而在移植术后,50%-70%的患者出现复发。
SGB-9768 是圣因生物自主研发的靶向补体C3的创新型小干扰RNA(siRNA)药物,采用GalNAc肝靶向递送技术,通过RNAi机制特异性抑制C3表达,从而从源头抑制补体系统的过度活化。
(5)勃林格殷格翰肺纤维化创新疗法那米司特片在华获批
10月22日,勃林格殷格翰宣布,其肺纤维化创新疗法口服选择性磷酸二酯酶4B(PDE4B)抑制剂博优维®(通用名:那米司特片)正式获得中国国家药品监督管理局(NMPA)批准,用于治疗成人特发性肺纤维化(Idiopathic Pulmonary Fibrosis,IPF)。
这是十年来首个在III期临床试验中达到主要终点并成功获批的IPF治疗药物,打破了该领域十余年无新药获批的僵局,标志着IPF治疗领域的突破性进展。
值得一提的是,中国实现了博优维®全球同步研发、同步注册、同步获批(仅在美国获批后2周内),真正实现了零时差惠及中国患者。此次在华获批是基于博优维®关键性III期临床试验FIBRONEER™-IPF的积极结果,该试验是IPF治疗领域迄今为止规模最大的III期临床研究。结果显示:与安慰剂相比,博优维®治疗组的患者在第52周时用力肺活量(FVC, ml)较基线的下降显著减少,达到了主要终点。
(6)首药控股1类新药索特替尼片申报上市
10月22日,首药控股完全自主研发的索特替尼片(SY-5007,商品名:首亦泽®)新药上市申请已获受理。该药物用于治疗RET基因融合阳性的局部晚期或转移性非小细胞肺癌成人患者,是首个进入临床研究,也是临床进展最快的完全国产的选择性RET抑制剂之一。
索特替尼片是首药控股自主研发的高选择性小分子RET酪氨酸激酶抑制剂。RET基因融合在非小细胞肺癌患者中的发生率约为1%-2%,这类患者确诊时多处于晚期阶段,传统化疗方案疗效有限且持续时间短,免疫疗法响应率低。关键性Ⅱ期和Ⅲ期临床试验数据显示,索特替尼在初治及经治患者中均展现出“起效迅速、缓解率高、持续时间长”的疗效特征。
目前国内仅有两款高选择性RET抑制剂获批,分别为礼来的塞普替尼、基石药业引进的普拉替尼。这两款药物均属于进口或引进药物,索特替尼有望成为首个国产自主研发的高选择性RET抑制剂。
(7)翰森制药新型RET抑制剂HS-10365胶囊申报上市
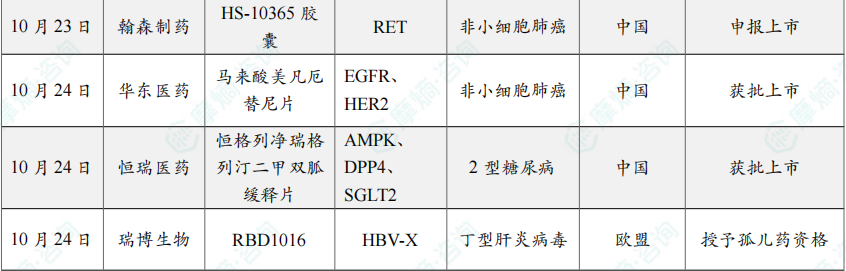
10月23日,国家药品监督管理局(NMPA)药品审评中心(CDE)官网显示,翰森制药的1类新药HS-10365胶囊的上市申请已获受理。
HS-10365是一款高活性、高选择性的RET酪氨酸激酶抑制剂,此前I期临床研究结果显示,该药在RET融合阳性非小细胞肺癌(NSCLC)患者中展现出令人鼓舞的抗肿瘤活性与持久缓解,且安全性可控,PK特性良好。
翰森目前正在开展HS-10365一线治疗RET融合阳性局部晚期或转移性非小细胞肺癌的有效性和安全性的注册性临床研究。
(8)华东医药EGFR/HER2小分子抑制剂马来酸美凡厄替尼片获批上市
10月24日,国家药监局(NMPA)官网显示,华东医药的马来酸美凡厄替尼片(曾用名:迈华替尼)获批上市,用于表皮生长因子受体(EGFR)21号外显子L858R置换突变的局部晚期或转移性非小细胞肺癌(NSCLC)成人患者的一线治疗一线治疗。
马来酸美凡厄替尼片为一款新型、强效、高选择性、具有口服活性的不可逆EGFR/人表皮生长因子受体-2(HER2)小分子抑制剂,是全新的、拥有自主知识产权的国家1类创新药。该药通过与EGFR(ErbB1)和HER2(ErbB2)的激酶区域共价结合,不可逆地抑制酪氨酸激酶自磷酸化,导致ErbB信号下调,从而抑制肿瘤生长。
2015年,中美华东与苏州迈泰生物技术有限公司和杭州华东医药集团新药研究院有限公司签订协议,取得马来酸美凡厄替尼原料药及其制剂在中国市场的权益,并在约定区域内进行产品新药技术开发和商业化的权利。
(9)恒瑞医药三合一口服降糖药恒格列净瑞格列汀二甲双胍缓释片获批上市
10月24日,国家药品监督管理局(NMPA)官网显示,恒瑞医药的恒格列净瑞格列汀二甲双胍缓释片(HR20031片)的上市申请已获批准,用于经二甲双胍治疗后血糖仍控制不佳的2型糖尿病患者。
HR20031片是恒瑞医药自主研发的钠-葡萄糖协同转运体2(SGLT2)抑制剂恒格列净、二肽基肽酶-4(DPP-4)抑制剂磷酸瑞格列汀和二甲双胍的固定剂量复方缓释制剂,通过三种不同作用机制达到降血糖作用,拟每日1次口服用于治疗经二甲双胍治疗后血糖仍控制不佳的2型糖尿病,以改善此类患者的血糖控制。
2023年6月,该三药联合的III期临床试验(SHR3824-SP2086-MET-301)主要研究终点达到方案预设的优效标准。研究结果表明,三药联合剂量组分别优效于两药联合剂量组,即24周糖化血红蛋白(HbA1c)相对基线下降的组间差值均具有统计学显著差异和临床意义的改变。同时,三药联合剂量组安全性、耐受性良好。
(10)瑞博生物治疗丁肝病毒感染的小核酸药物RBD1016获EMA孤儿药资格
10月24日,瑞博生物及其子公司Ribocure Pharmaceuticals共同宣布欧洲药品管理局(EMA)授予其小干扰RNA(siRNA)候选药物 RBD1016 孤儿药资格(ODD),用于治疗丁型肝炎病毒(HDV)的感染。
瑞博生物新闻稿介绍,其自主研发的RiboGalSTAR肝靶向递送平台的安全性、有效性和长效性已通过多项临床研究得以验证,其中包括用于治疗HDV适应症的 RBD1016,目前该药物正在全球同步推进乙肝和丁肝的2期临床试验。
HDV是病毒性肝炎中最严重的类型,仅感染已患有乙型肝炎(HBV)的患者。HDV会加速肝病进展,显著增加肝硬化、肝衰竭和肝癌的风险。瑞博生物的 RBD1016 小核酸药物精准靶向HDV病毒,有望为患者提供更有效、更持久的治疗方案。
1.4.2 本周全球TOP10积极/失败临床结果
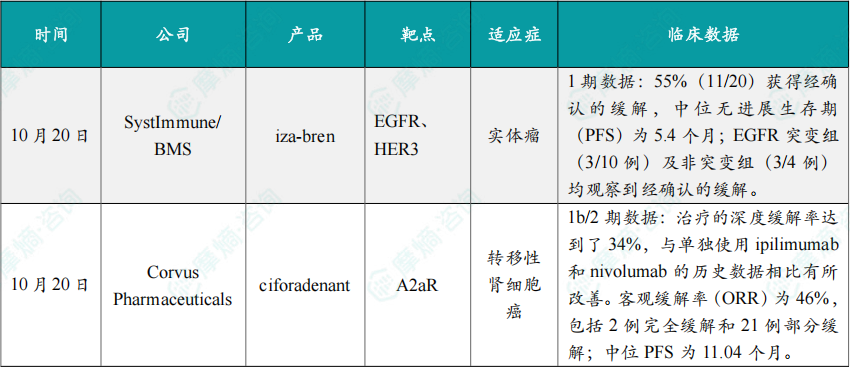
(1)SystImmune/BMS公布iza-bren 1期临床试验数据
10月20日,SystImmune公司与百时美施贵宝公布其全球1期研究 US-Lung-101 的安全性和有效性数据。该研究评估了ADC疗法 iza-bren 在多种肿瘤患者中的疗效与安全性。
Iza-bren 是一款潜在“first-in-class”的EGFR/HER3靶向双特异性ADC。该疗法的双重作用机制可阻断EGFR和/或HER3信号,抑制癌细胞增殖与存活,同时在抗体介导的内化后释放Topo1i有效载荷,引发细胞毒性反应,最终导致癌细胞死亡。Iza-bren 已获美国FDA授予突破性疗法认定,用于既往接受过治疗的EGFR突变型非小细胞肺癌(NSCLC)患者。
截至2025年7月23日,iza-bren 在多种高度预处理的实体瘤患者中,包括EGFR突变型与野生型NSCLC,展现出令人鼓舞的抗肿瘤活性,并具备可控的安全性特征。血液学不良事件可通过常规医学手段有效控制,且未观察到间质性肺病。其中,接受2.5 mg/kg剂量治疗的患者中,55%(11/20)获得经确认的缓解,中位无进展生存期(PFS)为5.4个月;EGFR突变组(3/10例)及非突变组(3/4例)均观察到经确认的缓解。
(2)Corvus Pharmaceuticals公布ciforadenant 1b/2期临床试验中期数据
10月20日,Corvus Pharmaceuticals公司宣布,其候选药物 ciforadenant 用于治疗转移性肾细胞癌(RCC)患者的1b/2期临床试验取得了积极的中期数据。
Ciforadenant 是一种在研口服小分子免疫检查点抑制剂。此前,ciforadenant 已被证明可以阻断肿瘤中髓系细胞的免疫抑制作用。临床前研究结果显示,ciforadenant 与抗PD-1和抗CTLA-4抗体联合使用具有协同作用。
截至2025年5月的数据,ciforadenant 与 ipilimumab 和 nivolumab 的三药联合疗法是可行且耐受性良好的。在年龄中位数为61.5岁的患者群体中(其中82%为预后较差或中等的患者),治疗的深度缓解率达到了34%,与单独使用 ipilimumab 和 nivolumab 的历史数据相比有所改善。客观缓解率(ORR)为46%,包括2例完全缓解和21例部分缓解;中位PFS为11.04个月。
(3)Moderna公布癌症疫苗mRNA-4359联合疗法1/2期临床试验初步数据
10月20日,Moderna公司宣布,其癌症疫苗 mRNA-4359 联合PD-1抑制剂 pembrolizumab,用于治疗免疫检查点抑制剂耐药或复发(CPI-R/R)黑色素瘤患者的1/2期临床研究获得积极初步结果。
mRNA-4359 是一种在研的免疫逃逸靶向癌症抗原疗法,可编码PD-L1与IDO1两种常见免疫逃逸通路的表位,以诱导抗原特异性T细胞反应,从而直接杀伤肿瘤细胞并清除免疫抑制细胞。
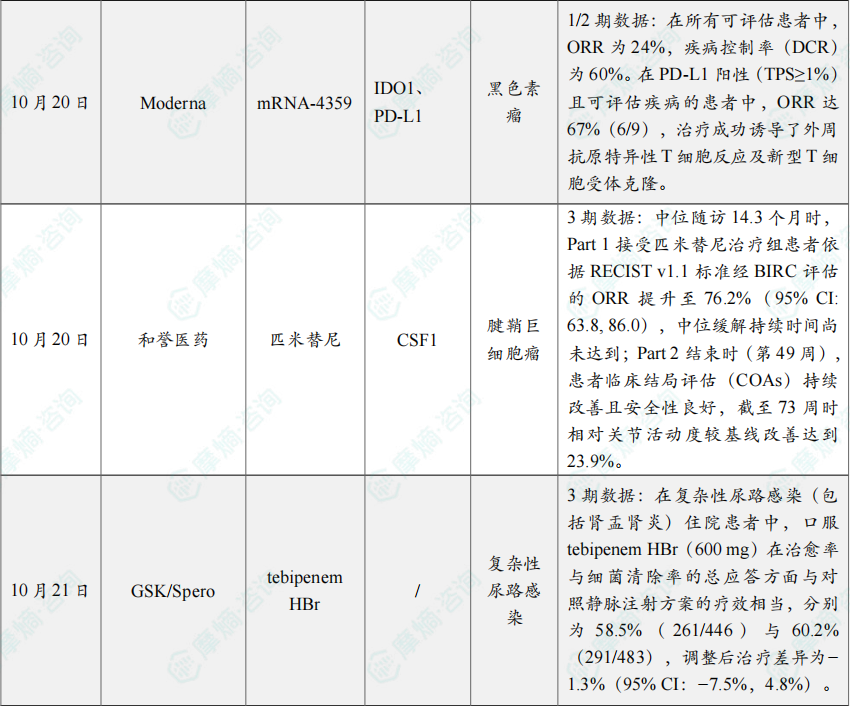
研究纳入29例曾接受至少一种免疫检查点抑制剂治疗的CPI-R/R黑色素瘤患者。结果显示,在所有可评估患者中,ORR为24%,疾病控制率(DCR)为60%。在PD-L1阳性(TPS≥1%)且可评估疾病的患者中,ORR达67%(6/9),治疗成功诱导了外周抗原特异性T细胞反应及新型T细胞受体克隆。研究中尚未达到中位缓解持续时间(DOR),提示该疫苗的疗效持久。mRNA-4359 目前已进入该1/2期研究的临床2期部分。
(4)和誉医药公布匹米替尼临床3期MANEUVER研究长期疗效和安全性数据
10月20日,和誉医药宣布,公司在2025年欧洲肿瘤内科学会年会(ESMO Congress 2025)上以口头报告形式展示了其自主研发的小分子CSF-1R抑制剂 匹米替尼(pimicotinib / ABSK021)治疗腱鞘巨细胞瘤(TGCT)患者的全球III期MANEUVER研究的长期疗效和安全性数据。结果显示,依据RECIST v1.1标准经盲态独立评审委员会(BIRC)评估,匹米替尼在TGCT患者中展现出强劲而持久的肿瘤缓解疗效,临床结局评估(包括关节活动度、僵硬、疼痛和身体功能)表现出具有临床意义的持续改善,安全性与既往分析一致,其应用于TGCT长期治疗的可行性得到进一步验证。
匹米替尼是由和誉医药独立研发的一款新型、口服、高选择性且高效的小分子CSF-1R抑制剂,其全球III期MANEUVER研究由三部分构成,旨在评估匹米替尼在TGCT患者中的疗效和安全性。
本届ESMO年会上,MANUEVER研究Leading PI,北京积水潭医院牛晓辉教授现场解读了本研究的长期疗效和安全性数据:中位随访14.3个月时,Part 1接受匹米替尼治疗组患者依据RECIST v1.1标准经BIRC评估的ORR提升至76.2%(95% CI: 63.8, 86.0),中位缓解持续时间尚未达到;Part 2结束时(第49周),患者临床结局评估(COAs)持续改善且安全性良好,截至73周时相对关节活动度较基线改善达到23.9%。对照组患者改用匹米替尼后,同样获得临床获益,中位随访时间8.5个月时,ORR达到64.5%,且COAs也有所改善。
(5)GSK/Spero公布口服碳青霉烯类抗生素tebipenem HBr关键3期临床结果
10月21日,GSK与Spero Therapeutics联合公布了关键性3期PIVOT-PO研究的完整积极结果。该研究评估了口服在研药物 tebipenem HBr 用于治疗复杂性尿路感染(cUTI),包括肾盂肾炎的疗效与安全性。
研究结果显示,tebipenem HBr 达到了主要终点,与静脉给药方案相比表现出非劣效性。相关数据将提交至监管机构,以支持后续的上市申请。该试验于5月因疗效显著而提前终止。
分析显示,在复杂性尿路感染(包括肾盂肾炎)住院患者中,口服 tebipenem HBr(600 mg)在治愈率与细菌清除率的总应答方面与对照静脉注射方案的疗效相当,分别为58.5%(261/446)与60.2%(291/483),调整后治疗差异为−1.3%(95% CI:−7.5%,4.8%)。
安全性方面,tebipenem HBr 与对照静脉注射方案及其他碳青霉烯类抗生素总体一致,最常见的不良事件(≥3%)为腹泻和头痛,均为轻中度、非严重事件。
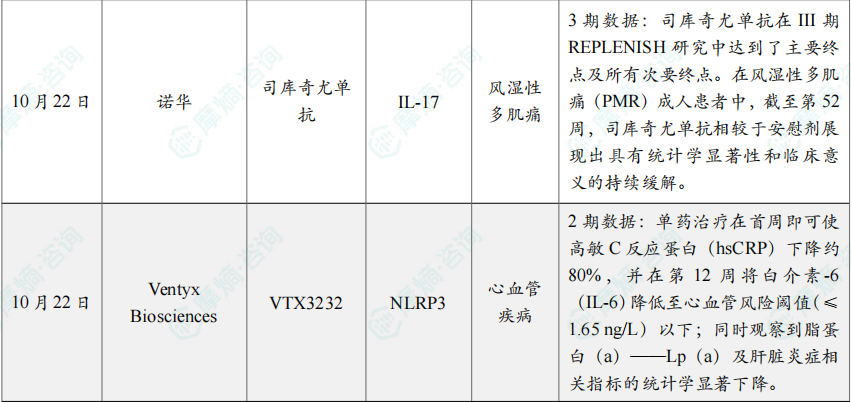
(6)诺华的司库奇尤单抗治疗风湿性多肌痛3期研究成功
10月22日,诺华宣布,司库奇尤单抗在III期REPLENISH研究中达到了主要终点及所有次要终点。
在风湿性多肌痛(PMR)成人患者中,截至第52周,司库奇尤单抗相较于安慰剂展现出具有统计学显著性和临床意义的持续缓解。
REPLENISH是一项全球多中心、随机、双盲、安慰剂对照、平行组设计的III期临床研究(NCT05767034),旨在评估司库奇尤单抗治疗PMR患者的疗效与安全性。
患者被随机分配至三个治疗组:司库奇尤单抗300mg组、司库奇尤单抗150mg组及安慰剂组,所有治疗组均联合24周糖皮质激素逐渐减量方案。该研究的主要终点是,在第52周时实现持续缓解的患者比例。关键次要终点包括:第52周时实现完全持续缓解的患者比例、校正后年度累积糖皮质激素剂量,以及截至第52周时首次使用补救治疗(escape/rescue treatment)的时间。其详细数据将在即将召开的医学大会上公布,新适应症申请将于2026年上半年提交至各国监管机构。
(7)Ventyx Biosciences的NLRP3抑制剂VTX3232临床2期研究成功
10月22日,Ventyx Biosciences宣布,其口服、每日一次的小分子 VTX3232 在一项针对肥胖并伴心血管风险因素患者的2期研究中取得积极结果。
该研究共入组175例受试者,比较 VTX3232 与安慰剂在疗效与安全性方面的差异;结果显示,VTX3232 无论作为单药还是与司美格鲁肽联用,均表现出良好的安全性和耐受性。
进一步分析显示,VTX3232 可显著降低多项心血管风险因素:单药治疗在首周即可使高敏C反应蛋白(hsCRP)下降约80%,并在第12周将白介素-6(IL-6)降低至心血管风险阈值(≤1.65 ng/L)以下;同时观察到脂蛋白(a)——Lp(a)及肝脏炎症相关指标的统计学显著下降。与司美格鲁肽联用可带来额外获益,但无论单药或联用,VTX3232 对体重均未产生影响。
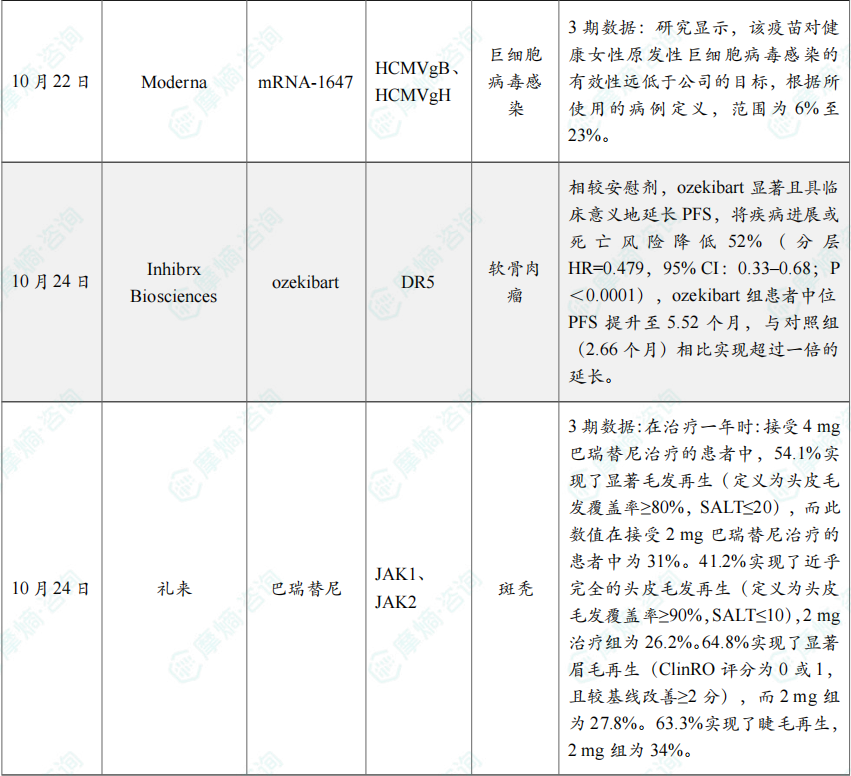
(8)Moderna巨细胞病毒疫苗mRNA-1647 3期临床失败
10月22日,Moderna宣布其备受关注的巨细胞病毒(CMV)疫苗在三期临床试验中未能达到主要疗效终点,决定终止针对先天性CMV的疫苗研发计划。不过,公司表示将继续探索该疫苗在其他适应症中的潜力。
根据新闻稿,该三期临床试验(NCT05085366)评估了其研究性巨细胞病毒疫苗mRNA-1647的疗效,但该研究未能达到主要疗效终点——预防血清阴性育龄女性(16-40岁)的CMV感染。
Moderna的该试验是一项随机、观察者盲法、安慰剂对照研究。试验共纳入了来自13个国家约300个中心的约7500名16-40岁女性,是迄今为止评估CMV疫苗疗效的最大规模试验。研究显示,该疫苗对健康女性原发性巨细胞病毒感染的有效性远低于公司的目标,根据所使用的病例定义,范围为6%至23%。由于试验结果未达预期,Moderna决定终止临床开发计划。
(9)Inhibrx Biosciences软骨肉瘤创新单抗ozekibart临床试验成功
10月24日,Inhibrx Biosciences宣布,其注册性ChonDRAgon研究(n=206)在晚期或转移性、不可切除性软骨肉瘤患者中取得积极的无进展生存期(PFS)结果。该试验评估 ozekibart(INBRX-109)单药对比安慰剂的疗效与安全性。
根据新闻稿,ozekibart 为首个在随机对照试验中于软骨肉瘤显示显著PFS获益的在研疗法,而该疾病目前尚无获批的系统性治疗选择。基于上述结果,公司计划于2026年第二季度提交生物制品许可申请(BLA)。
Ozekibart 为四价DR5激动型抗体,旨在通过DR5激活诱导肿瘤细胞死亡。研究达到主要终点:相较安慰剂,ozekibart 显著且具临床意义地延长PFS,将疾病进展或死亡风险降低52%(分层HR=0.479,95% CI:0.33–0.68;P<0.0001),ozekibart 组患者中位PFS提升至5.52个月,与对照组(2.66个月)相比实现超过一倍的延长。安全性方面,ozekibart 总体耐受性良好。
(10)礼来斑秃新药巴瑞替尼3期结果亮眼
10月24日,礼来(Eli Lilly and Company)公布了其与Incyte联合开发的JAK抑制剂 Olumiant(baricitinib,巴瑞替尼),治疗重度斑秃(AA)青少年患者(12至<18岁)的3期临床试验BRAVE-AA-PEDS的最新结果。
分析显示,患者在接受治疗一年后实现头皮、眉毛及睫毛的显著再生。礼来计划向全球监管机构提交此数据,以推动该疗法适应症标签更新,并计划于明年启动BRAVE-AA-PEDS试验的6至<12岁儿童患者入组。分析显示,在治疗一年时:接受4 mg巴瑞替尼治疗的患者中,54.1%实现了显著毛发再生(定义为头皮毛发覆盖率≥80%,SALT≤20),而此数值在接受2 mg巴瑞替尼治疗的患者中为31%。41.2%实现了近乎完全的头皮毛发再生(定义为头皮毛发覆盖率≥90%,SALT≤10),2 mg治疗组为26.2%。64.8%实现了显著眉毛再生(ClinRO评分为0或1,且较基线改善≥2分),而2 mg组为27.8%。63.3%实现了睫毛再生,2 mg组为34%。在重度疾病患者(基线SALT评分50-94)中,4 mg组有71%实现了显著毛发再生,2 mg组为58.6%。
同期事件:
1. 2025年第43周10.20-10.26国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2025年第43周10.20-10.26国内仿制药/生物类似物申报/审批数据分析
3. 2025年第43周10.20-10.26国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2025.10.20-2025.10.26) },如需查看或下载完整版报告,可点击!
扩展阅读:
1. 2025年第17周04.21-04.27全球创新药研发概览
2. 2025年第16周04.14-04.20全球创新药研发概览
3. 2025年第11周03.10-03.16全球创新药研发概览
4. 2025年第7周02.10-02.16全球创新药研发概览
5. 2025年第1周12.30-01.05全球创新药研发概览
想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论