
































摘要:
1000亿美元。
在 立普妥(Lipitor)出现之前,这是一个制药行业无法想象的天文数字。此前,一款药物年销售额达到10亿美元就被称为“重磅炸弹”(Blockbuster);而立普妥,硬生生将这个天花板捅破,重新定义了商业药物的极限。
它是如何做到的?
脂质假说与胆固醇的生化迷宫:从病理发现到靶点确立
1. 胆固醇发现、研究及药物研发历程
胆固醇(Cholesterol),这一如今被公众视为心血管健康“头号大敌”的物质,其科学发现史可追溯至18世纪。
- 1784年:法国医生François Poulletier de la Salle首次在胆结石中发现了胆固醇。
- 1841年:俄国科学家Vogel证实动脉血管壁的粥样硬化斑块中存在胆固醇。
- 1910年:德国化学家Adolf Windaus(“类固醇之父”)在研究人体解剖标本时,最早揭示了动脉粥样硬化与胆固醇异常升高之间的关联。
- 1913年:俄国病理学家Anitschkow通过喂食家兔高胆固醇饮食,成功构建了世界上首个动脉粥样硬化动物模型,侧面验证了Adolf Windaus的发现。
- 20世纪50年代(起):生物化学家Huff及其团队开始投身于胆固醇生物合成路径的探索工作。
- 1956年:Huff团队成功从酵母提取物中分离出甲羟戊酸(MVA),并证实MVA是胆固醇合成过程中的关键中间体。
- 1959年:德国Max-Planck研究所率先发现了HMG-CoA还原酶。
- 20世纪60年代
科学研究进展: Siperstein等科研人员证明HMG-CoA还原酶在胆固醇合成代谢中发挥着核心催化作用(促使HMG-CoA转化为MVA),抑制该酶活性可显著降低血浆胆固醇水平。
社会健康背景: 冠心病成为美国人群的首要死因。当时市面上的降脂药物(如氯贝特、烟酸、胆胺)在安全性和有效性上存在不足,临床急需更安全有效的降脂方法。
- 20世纪70年代起:鉴于HMG-CoA还原酶作为药物靶点的优势(底物水溶性、无累积毒性),研发HMG-CoA还原酶抑制剂逐渐成为新型心血管疾病治疗药物的重要研究方向。
2. 生命的双刃剑:生理功能与病理机制
在深入探讨药物机制之前,必须客观认识胆固醇在生物学上的核心地位。
胆固醇并非单纯的毒素,而是真核细胞膜流动性的关键调节者,是维持细胞完整性的“水泥”。此外,它还是合成维生素D、胆汁酸以及皮质醇、醛固酮、性激素等甾体激素的通用前体。人体内约70%-80%的胆固醇由肝脏内源性合成,仅有20%-30%源自外源性食物摄入。
胆固醇具有疏水性,无法直接溶于水,故而需与低密度脂蛋白(Low Density Lipoprotein,LDL)以及高密度脂蛋白(High Density Lipoprotein,HDL)相结合,方能随血液循环运输至全身各处。
- 低密度脂蛋白(LDL):负责将胆固醇从肝脏运往外周组织。当其水平过高时,易在受损的血管内皮下氧化沉积,被巨噬细胞吞噬后形成“泡沫细胞”,这是动脉粥样斑块的核心成分,因此被称为“坏胆固醇”。
- 高密度脂蛋白(HDL):执行“逆向转运”功能,将外周组织的胆固醇回收至肝脏进行代谢,被称为“好胆固醇”。
当LDL与HDL的平衡被打破,过剩的胆固醇便在血管壁堆积,导致管腔狭窄、血流受阻,最终可能诱发急性心肌梗死或脑卒中。20世纪60年代,随着流行病学数据的积累,高胆固醇血症被确认为冠心病的主要风险因素,寻找能够有效降低血浆胆固醇的药物成为了医药界的圣杯。
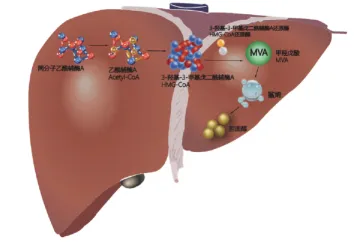
3. 关键限速步:甲羟戊酸途径与HMG-CoA还原酶
要阻断胆固醇的合成,必须从其生化合成路径入手。20世纪50年代,生物化学家们逐步绘制出了胆固醇合成的精细路线图——甲羟戊酸途径。这一途径涉及30多步酶促反应,但真正的控制阀门位于上游:
乙酰辅酶A首先缩合生成3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)。随后,在HMG-CoA还原酶的催化下,HMG-CoA消耗NADPH还原为甲羟戊酸(Mevalonate, MVA)。
这一步是整个合成路径的限速步骤,也是负反馈调节的主要位点。
一旦甲羟戊酸生成,后续转化为鲨烯(Squalene)、羊毛固醇(Lanosterol)直至胆固醇的过程便不可逆转。因此,HMG-CoA还原酶成为了理想的药物靶点:抑制该酶不仅能高效阻断胆固醇合成,且底物HMG-CoA水溶性好、易代谢,不会造成中间产物毒性堆积。
胆固醇的生物合成途径
他汀前传:从真菌代谢物到第一代药物的竞争
1. 远藤章的执念与青霉素的启示
历史总是惊人的相似。正如弗莱明从霉菌中发现了青霉素,日本三共制药(Sankyo)的远藤章(Akira Endo)也坚信真菌中藏着抑制胆固醇合成的秘密。作为“类固醇之父”的信徒,远藤章推测微生物为了在生态位竞争中生存,可能会分泌某种物质来抑制其他生物合成固醇(细胞膜的重要成分),从而杀死竞争对手。
1971年起,远藤章团队筛选了6000多种真菌发酵液。1973年,他们终于成功分离出HMG-CoA还原酶的抑制剂——美伐他汀(Mevastatin,ML-236B),此乃全球首个他汀类化合物。体外实验显示其抑制活性极高,但在大鼠实验中却遭遇滑铁卢——大鼠血脂未见下降。这一度让项目濒临崩溃,直到远藤章在三共制药一位研究员的帮助下,在产蛋母鸡身上重复了实验(母鸡的胆固醇代谢模式更接近人类),才观察到了惊人的降脂效果(两周下降34%)。
然而,命运多舛。1980年,一项为期两年的犬长期毒理试验显示,美伐他汀可能增加淋巴瘤发病风险。三共制药被迫终止了美伐他汀的开发。
2. 默沙东的崛起与“他汀战争”
在大洋彼岸,制药巨头默沙东在阿尔弗雷德·阿尔伯茨(Alfred Alberts)的带领下,利用其强大的微生物发酵库,从土曲霉中分离出了结构极其相似的化合物——洛伐他汀(Lovastatin)。
由于洛伐他汀与美伐他汀作用机理一致且结构相似,这意味着洛伐他汀也极有可能引发淋巴瘤。于是,默沙东公司决定暂停临床试验,开展大规模动物毒理学实验。经过两年的毒理学研究,未发现任何确切的致癌证据,默沙东公司遂决定重启临床试验。
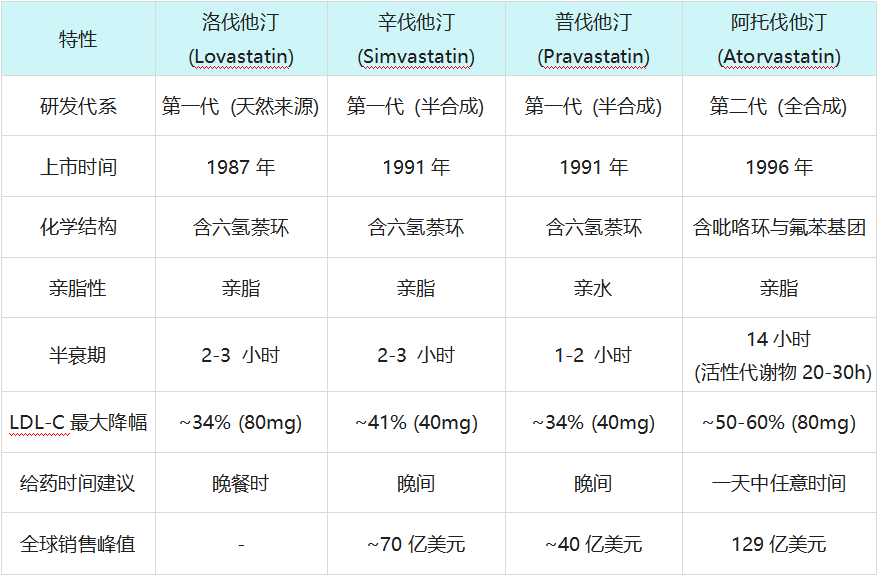
1987年,历经漫长审查,洛伐他汀(商品名:Mevacor)获批上市,这成为全球首款上市的他汀类药物,标志着人类正式进入“他汀时代”。紧随其后,默沙东通过化学修饰开发了半合成的辛伐他汀(Simvastatin),三共制药推出了普伐他汀(Pravastatin),山德士推出了首个全合成他汀——氟伐他汀(Fluvastatin)。
截至1990年代中期,他汀市场已是群雄逐鹿。第一代他汀(如洛伐他汀、辛伐他汀)多为真菌来源或半合成衍生物,具有六氢萘环结构,虽然有效,但在药效强度和组织选择性上仍有提升空间。
阿托伐他汀的诞生:帕克-戴维斯的逆袭
1. Bruce Roth的分子雕刻术
1981年,年仅27岁的Bruce Roth于罗切斯特大学化学系投身他汀类化合物的全合成研究工作。1982年,他加入了帕克-戴维斯公司(华纳-兰伯特公司的分公司)。基于此前的研究积累,Roth极力推荐公司启动他汀类药物研发项目。彼时,默沙东的洛伐他汀尚未上市,但研发竞赛已然白热化。Roth的目标不仅仅是模仿,而是超越。他致力于开发全合成的“II型他汀”,试图摆脱天然产物的结构限制。
Roth团队在设计阿托伐他汀时,引入了一个独特的五元吡咯环作为核心骨架,并创造性地附加了一个氟苯基团。这种结构设计并非随意为之,而是基于对酶活性位点的深刻理解。氟苯基团能够与HMG-CoA还原酶活性口袋中的特定疏水区域形成更强的相互作用,赋予了阿托伐他汀比天然他汀更强的亲和力。
1985年,Roth团队成功首次合成阿托伐他汀分子。
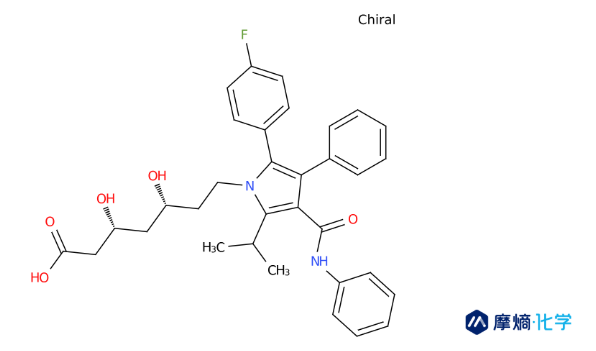
2. 拆分还是消旋?一个价值千亿的抉择
合成成功只是第一步,Roth面临着一个巨大的工艺挑战:手性。阿托伐他汀分子含有两个手性中心,合成产物通常是消旋体(由两种镜像异构体混合而成)。
当时的竞争对手山德士选择了以消旋体形式开发氟伐他汀。然而,生物体内酶的识别具有高度的立体专一性,通常只有一种构型有效,另一种无效异构体(Distomer)不仅是代谢负担,还可能引发未知的毒性。
帕克-戴维斯的高层面临艰难抉择:是开发消旋体以节省时间和成本,还是死磕单一异构体?
- 消旋体路径:合成简单,成本低,但他汀类药物只有一半有效,FDA极有可能拒绝批准。
- 单一异构体路径:需要复杂的手性合成工艺,耗时耗资,但能获得活性更高的纯净药物。
最终,出于对“纯净科学”的坚持以及规避潜在毒性的考量,公司决定开发纯光学异构体(R,R-型)。这一决定导致工艺开发耗时两年多,但也正是这一高纯度的特性,为后来阿托伐他汀展现出的卓越疗效和安全性奠定了物质基础。如果当时选择了消旋体,阿托伐他汀可能沦为平庸之辈,绝无后来“药王”的风采。
3. Roger Newton的“惊天一跪”
1989年,阿托伐他汀终于完成了临床前研究。此时,市场上已有三款他汀(洛伐他汀、辛伐他汀、普伐他汀)上市,山德士的氟伐他汀也即将获批。作为“第五个出场”的选手,阿托伐他汀似乎注定只能喝点残汤剩水。
更糟糕的是,华纳-兰伯特公司内部因业绩下滑正在进行大规模裁员,管理层计划砍掉“拥挤赛道”中的阿托伐他汀项目,以节省开支。
在那场决定命运的会议上,Roger Newton和Bruce Roth面对的是充满怀疑的高管。为了挽救这个被视为“多余”的项目,项目药理学负责人Newton据传甚至在会议室单膝下跪,恳请公司给这个药物一次进入临床的机会。他列举了动物实验中的关键数据:
- 肝脏高选择性:药物主要集中在肝脏发挥作用,外周副作用可能更低。
- 长半衰期:在大鼠和狗身上表现出远超其他他汀的半衰期。
- 强效性:在动物模型中,低剂量即可显著降低甘油三酯,这是其他他汀不具备的优势。
最终,研发总裁Ronnie Cresswell被打动,阿托伐他汀惊险地拿到了通往临床试验的入场券。这一跪,跪出了未来1250亿美元的销售神话。
绝地反击:“All or Nothing”的临床豪赌
1. 药代动力学的惊喜:全天候护盾
I期临床试验结果令人震惊且兴奋。阿托伐他汀不仅耐受性良好,其血浆半衰期更是长达14小时,远超同类药物(通常为1-3小时)。更独特的是,它的代谢产物同样具有抑制酶的活性,这使得其在人体内对HMG-CoA还原酶的总抑制效应可持续20-30小时。
这意味着阿托伐他汀可以全天候地压制胆固醇合成,而不仅仅是在夜间(人体胆固醇合成的高峰期)。相比之下,半衰期短的他汀通常建议在睡前服用,而阿托伐他汀可以在一天中任何时间服用,这极大地提高了患者的依从性。
2. 定义“超级他汀”的CURVES研究
面对市场上根深蒂固的竞争对手,帕克-戴维斯公司负责临床研究的副总裁Black提出了“all or nothing”策略。
当时的逻辑是:如果阿托伐他汀只是“另一个他汀”,即便上市也无法在默沙东的阴影下生存。唯一的出路是证明它是“超级他汀”。
Black决定重新定义“剂量-效应”曲线。1994年启动的CURVES研究(Comparative dose efficacy study)是这一策略的完美执行。研究设计了一个直接的“头对头”(Head-to-Head)比拼:将10mg的阿托伐他汀与10mg、20mg、40mg甚至80mg的其他他汀进行对比。
结果是毁灭性的碾压:
- 10mg 阿托伐他汀(起始剂量)降低LDL-C的效果,超过了辛伐他汀40mg(当时的最高推荐剂量)以及其他所有他汀的最高剂量。
- 80mg 阿托伐他汀更是将LDL-C降低了近60%,这是当时任何药物都无法企及的深度。
这一结果在1994年蒙特利尔的科学会议上首次公布时,震惊了全场。据报道,默沙东的一位高管甚至站起来建议将其改名为“涡轮他汀”(Turbostatin),以此表达对其强效的敬畏。
此外,针对罕见病“纯合子家族性高胆固醇血症”(HoFH)的疗效,使得阿托伐他汀获得了FDA的快速通道审批资格。对于这种此前几乎无药可救的疾病,阿托伐他汀展现出了临床获益,这不仅是科学上的胜利,也为加速上市铺平了道路。
立普妥(阿托伐他汀)与早期他汀类药物的关键特性对比
商业帝国的构建:辉瑞入局与市场营销的艺术
1. 强强联手:华纳-兰伯特与辉瑞的联姻
1996年,阿托伐他汀正处于上市前夕。华纳-兰伯特虽然手握这一核武器,却发现自己缺乏足够的销售网络来引爆市场。彼时,辉瑞(Pfizer)正在积极寻找新产品以充实其管线,且拥有一支以激进和高效著称的销售铁军。
双方一拍即合,签署了共同推广协议。辉瑞庞大的医药代表军团与华纳-兰伯特的研发成果形成了完美互补。辉瑞在立普妥上市前就培训了超过2000名销售代表,确保在上市第一天就能覆盖全美所有的心血管医生。
2. 重新定位:从“治病”到“生活方式”
1997年,阿托伐他汀以商品名“立普妥”(Lipitor)在美国上市。恰逢FDA在1997年放宽了直接面向消费者(DTC)的广播电视广告限制。辉瑞敏锐地抓住了这一历史机遇,发起了铺天盖地的广告攻势。
他们的营销策略极具前瞻性:
- 扩大适应症认知:不再将立普妥局限于治疗严重的“高血脂症”患者,而是将其重新定义为一种维持健康的“生活方式药物”。
- 恐惧与希望并存:广告语潜移默化地传递着这样的信息——即使你感觉健康,看不见的胆固醇也可能在暗中破坏你的血管;通过服用立普妥,你可以将胆固醇控制在“理想”水平(<100 mg/dL)。
- 形象代言:使用健康的运动型中年男性形象,暗示服用立普妥是保持活力的秘诀。
这种策略极大地扩展了潜在用户群,将目标人群从几百万确诊患者扩大到了数千万关注健康的普通人,甚至引发了关于“过度医疗化”的伦理讨论,但在商业上无疑是巨大的成功。
3. 世纪并购:辉瑞的野心与吞噬
立普妥上市后的表现超出了所有人的预期,迅速成为华纳-兰伯特最核心的资产。然而,2000年,当华纳-兰伯特计划与另一家公司(American Home Products)合并时,辉瑞感到了巨大的威胁——如果华纳-兰伯特被第三方收购,辉瑞可能会失去立普妥的共同推广权或利润分成。
为了完全掌控立普妥这一“印钞机”,辉瑞发起了医药史上著名的恶意收购。尽管华纳-兰伯特试图反抗,但在辉瑞开出的900亿美元天价面前,股东们选择了妥协。2000年,辉瑞成功吞并华纳-兰伯特。这一并购不仅巩固了辉瑞的全球霸主地位,也将立普妥彻底刻上了辉瑞的烙印。
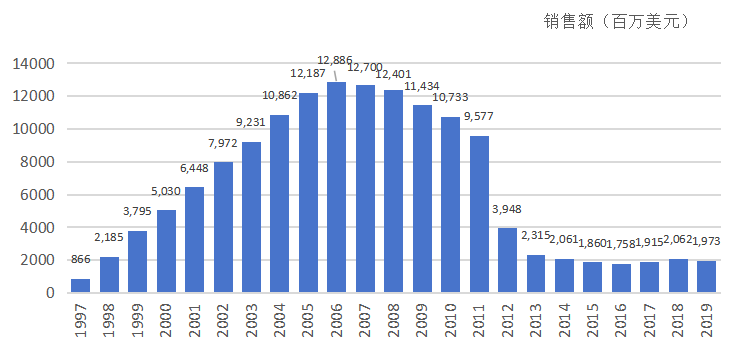
4. 专利保卫战与销售神话
随着销售额在2004年突破100亿美元,立普妥成为了仿制药厂商眼中的肥肉。印度仿制药巨头Ranbaxy在2004年发起了猛烈的专利挑战,试图证明辉瑞的关键专利(特别是针对单一异构体和晶型的专利)无效。
辉瑞组建了豪华律师团,利用阿托伐他汀独特的手性结构专利(R,R-异构体专利 US5273995)和后续申请的晶型专利构建了坚固的护城河。最终,双方达成和解,Ranbaxy的仿制药上市时间被推迟到2011年11月30日。这为立普妥赢得了宝贵的7年独占期。
在这期间,立普妥的年销售额连续7年超过100亿美元,并在2006年达到峰值128.86亿美元。截至2011年专利到期,立普妥累计销售额超过1250亿美元,创造了医药产品销售的辉煌业绩。
立普妥历年销售额
高胆固醇血症治疗药物的研究进展
据摩熵医药数据库的全球药物研发数据库显示,截至2025年1月2日,全球范围内处于研发进程中的高胆固醇血症治疗药物(涵盖创新药、改良型新药以及生物类似药)总计达131款。在这一庞大的研发阵列中,已有47款药物成功获批上市;另有3款药物已提交上市申请,正处于监管部门的审核流程中;处于三期临床试验阶段的药物数量为21款;处于一期临床试验与二期临床试验阶段的药物合计31款;尚处于临床前研究阶段的药物则有27款。
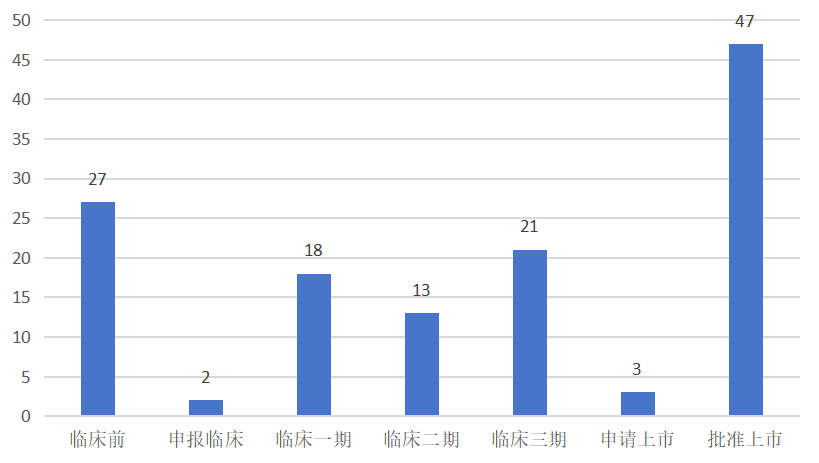
全球在研高胆固醇血症治疗药物开发阶段分布立普妥上市以后,截至2025年1月3日,全球范围内获批上市的创新药共计16款,具体信息如下表所示:
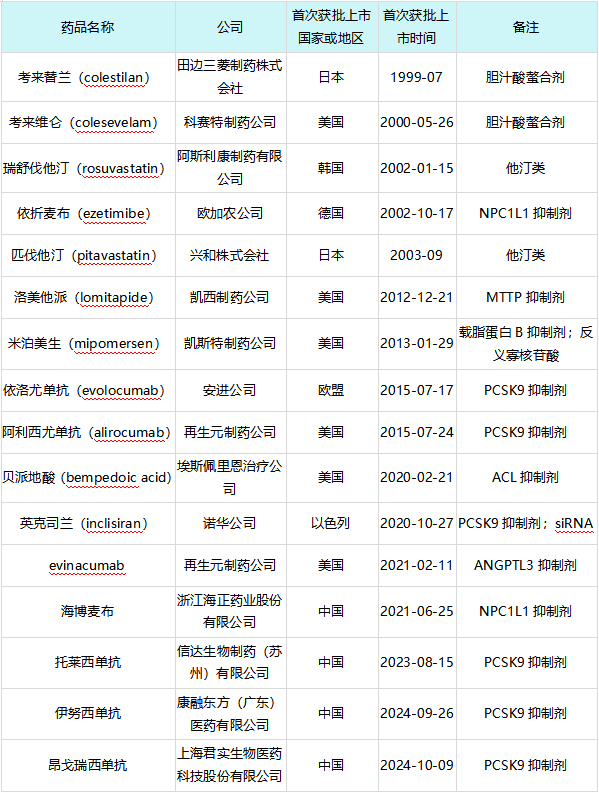
立普妥上市后全球范围内获批上市的高胆固醇血症创新药
1. 胆固醇吸收抑制剂:依折麦布(Ezetimibe)
如果说他汀是阻断了胆固醇的“生产线”,那么依折麦布则是封锁了“进口通道”。它通过抑制小肠刷状缘的NPC1L1蛋白,阻断外源性胆固醇的吸收。临床上常采用“他汀+依折麦布”的联合疗法,实现“1+1>2”的降脂效果。
2. 突破性的PCSK9抑制剂:受体循环的拯救者
在他汀之后,降脂领域最大的突破莫过于PCSK9抑制剂。
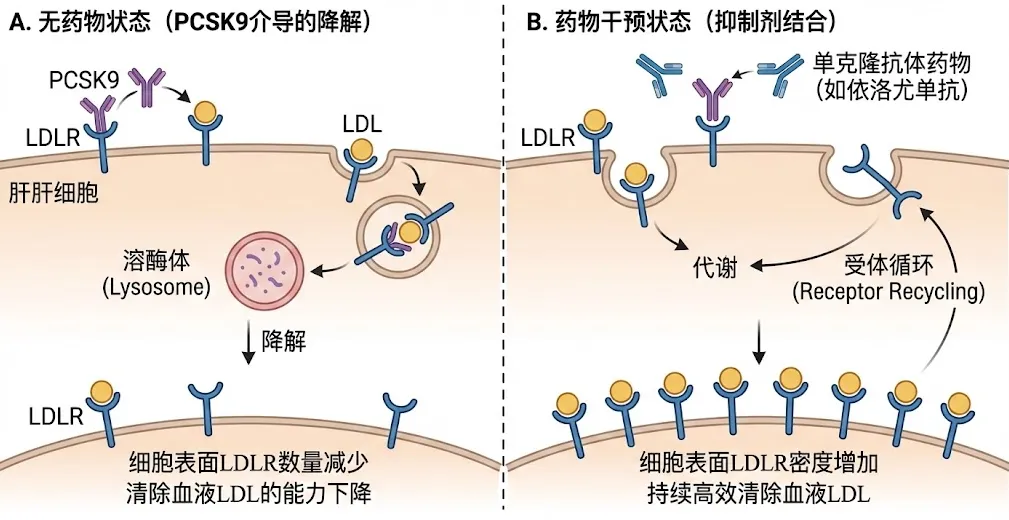
机制原理:肝细胞表面的LDL受体(LDLR)是清除血液中LDL的“清洁工”。正常情况下,LDLR在吞噬LDL后会返回细胞表面循环使用。然而,PCSK9(前蛋白转化酶枯草溶菌素/Kexin9型)蛋白会与LDLR结合,引导其进入溶酶体降解,阻止其循环。PCSK9就像是解雇清洁工的“监工”。
药物作用:依洛尤单抗(Evolocumab)和阿利西尤单抗(Alirocumab)是针对PCSK9的单克隆抗体。它们能精准结合血液中的PCSK9,阻止其与LDLR结合。这使得LDLR能够逃脱降解命运,重新回到肝细胞表面,继续高效清除血液中的LDL。临床数据显示,PCSK9抑制剂可在他汀基础上进一步降低LDL-C达50%-60%。
PCSK9抑制剂作用机制
然而,与立普妥时代“一超多强”的局面不同,后立普妥时代的竞争呈现出前所未有的内卷态势。根据摩熵医药靶点格局数据库,我们可以看到在全球PCSK9赛道上,不仅有国外老牌巨头,更涌现出大量中国力量。
诺华的英克司兰(Inclisiran)利用小干扰RNA(siRNA)技术,直接在肝细胞内降解PCSK9的mRNA,从源头阻断蛋白合成。其优势在于超长效,仅需每半年注射一次,被称为“降脂疫苗”。
3. 溯源阻断:ACL抑制剂
贝派地酸(Bempedoic acid)是一种ATP柠檬酸裂解酶(ACL)抑制剂。
机制原理:ACL位于HMG-CoA还原酶的上游,负责将线粒体输出的柠檬酸裂解为乙酰辅酶A(胆固醇合成的原料)。贝派地酸通过抑制ACL,切断了原料供应。
独特优势:它是一种前体药物,仅在肝脏中被特异性酶激活,而在肌肉组织中不激活。因此,它避免了他汀类药物常见的肌肉酸痛(肌病)副作用,为不耐受他汀的患者提供了绝佳选择。
4. 更多前沿靶点
- ANGPTL3抑制剂:Evinacumab,用于治疗纯合子家族性高胆固醇血症,通过解除ANGPTL3对脂蛋白脂酶的抑制,加速脂质代谢。
- Lp(a)抑制剂:针对脂蛋白(a)这一遗传性高风险因子的药物正在研发中,填补了这一领域的空白。
总结与反思
回顾立普妥的传奇,我们看到的不仅仅是分子的胜利,更是多维因素共振的结果。
- “同类最优”胜过“同类第一”
医药界常迷信“先发优势”(First-in-Class),认为第一个上市的药物将占据市场主导。然而,阿托伐他汀作为第五个上市的他汀类药物,彻底打破了这一教条。它证明了只要在疗效(Efficacy)和药代动力学(PK)特性上实现质的飞跃(Best-in-Class),后来者依然可以居上。罗斯对分子结构的精细打磨,特别是对单一异构体纯度的坚持,是产品力超越先发者的核心物理基础。
- 风险与收益的不对称博弈
帕克-戴维斯在CURVES研究中采用的“All or Nothing”策略,是基于深厚临床前数据的理性豪赌。这种敢于在试验设计中设定高门槛、以此换取高市场区分度的勇气,是现代药物开发中稀缺的品质。大多数公司倾向于设计“非劣效性”试验以求稳妥上市,而立普妥选择了“优效性”挑战,这种高风险带来了高回报。
- 科学家直觉与企业家的魄力
如果没有远藤章对真菌的执着,没有罗斯对合成化学的钻研,没有研究人员的执着,立普妥可能早已胎死腹中。而辉瑞通过资本运作将这一科学成果的商业价值放大到极致,则展示了现代医药工业中资本的雷霆手段。科学发现是火种,而资本和商业策略是将其引爆为燎原之势的助燃剂。
- 伦理的边界
立普妥的营销将药物从“治疗疾病”推向了“管理风险”甚至“改善生活方式”的范畴,这在造福数千万患者的同时,也引发了关于医疗资源分配和过度医疗的思考。当一项生理指标(如LDL-C)被定义为这就需要药物干预的“疾病”时,制药企业在公共卫生政策中的影响力不容小觑。
扩展阅读:
1. 已上市的新型降脂药inclisiran为什么可以长达6个月,一年仅用两次?
2. 诺华Q3财报解析:中国区净销售额增长18%,降脂药Leqvio翻倍增长!
3. 降脂新药SHR-1918:恒瑞医药国产首款ANGPTL3单抗III期研究正式启动
想要解锁更多药品信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论