

































药物代谢(生物转化)是机体抵御外界化学物质侵袭的核心防御机制。绝大多数临床药物具备较高脂溶性,必须依靠以肝脏为主的代谢酶系统,经历I相(功能化反应,如氧化、还原)和II相(结合反应,如葡萄糖醛酸化)的连续处理,转化为高水溶性产物从而跨越排泄屏障排出体外。
近年来,随着基因组学、系统生物学以及人工智能(AI)驱动的生理药代动力学(PBPK)模型的突破性进展,药物代谢研究已从传统的生化表征,跨越至动态、可预测的精准医学新纪元。当前的临床用药决策已不再局限于单一的代谢途径,而是全方位整合基因多态性、药物相互作用网络及微生态环境,为个体化精准给药提供科学锚点。
一、I相代谢酶系的生化重构与功能化网络
I相代谢的核心目的在于打破外源分子的化学惰性。这一时相不仅决定了药物的失活与清除速率,更与前药的体内活化及毒性反应中间体的生成密切相关。
1. 细胞色素P450超家族(CYP450s)的催化动力学特征
在众多I相代谢酶中,细胞色素P450超家族(CYP450s)无疑占据了统治地位。CYP450s属于含铁原卟啉IX辅基的血红素蛋白,广泛镶嵌于内质网的脂质双分子层中。其底物谱表现出极端的宽泛性,分子量跨度从简单的乙烯(28 Da)直至庞大的环孢素A(1201 Da)。
CYP450s本质上是一个复杂的混合功能单加氧酶系统。其催化循环高度依赖于烟酰胺腺嘌呤二核苷酸磷酸(NADPH)提供的电子池,并通过NADPH-细胞色素还原酶(以FAD和FMN为辅基)完成电子的跨膜接力传递。在这一生化反应中,CYP450s能够激活分子氧(O₂),将其中的一个氧原子精准插入底物分子内部,形成羟基化等氧化产物,而另一个氧原子则接受电子还原为水。值得注意的是,由于CYP450s活性中心的亚铁离子对一氧化碳(CO)表现出极高的亲和力,CO能够通过强烈的竞争性结合而全面抑制CYP450s介导的氧化代谢。
CYP450s 介导的药物氧化代谢
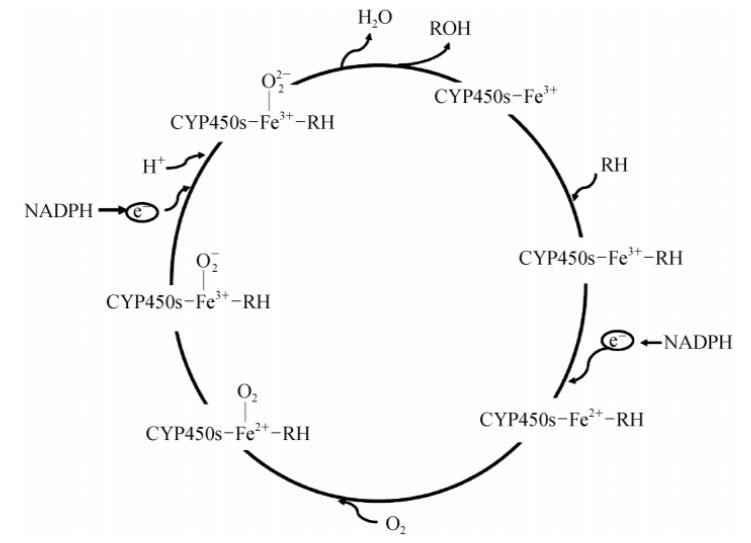
人体内参与药物代谢的CYP450s主要包括CYP1A2、CYP2C9、CYP2C19、CYP2D6和CYP3A等亚型,它们在组织分布、表达丰度及底物特异性上呈现出显著的异质性。其中,CYP3A家族(主要为CYP3A4和CYP3A5)表达最为丰富,占据了人肝微粒体CYP总量的约30%,并主导了临床上近50%药物的代谢。表1系统梳理了关键CYP450亚型的功能特征。
| 酶亚型 | 典型探针底物及特征性反应 | 代表性临床强效抑制剂 | 代表性药酶诱导剂 |
|---|---|---|---|
| CYP1A2 | 非那西丁 (O-去乙基化), 咖啡因 (N-去甲基化) | 呋拉茶碱、氟伏沙明、环丙沙星 | 吸烟、奥美拉唑、苯妥英钠 |
| CYP2C9 | S-华法林 (7-羟化), 甲苯磺丁脲 (甲基羟化) | 氟康唑、磺胺苯吡唑、胺碘酮 | 利福平、卡马西平、波生坦 |
| CYP2C19 | S-美芬妥因 (4'-羟化), 奥美拉唑 (5-羟化) | 噻氯匹定、氟康唑、苯环丙胺 | 利福平、青蒿素 |
| CYP2D6 | 右美沙芬 (O-去甲基化), 丁味洛尔 (1'-羟化) | 奎尼丁、氟西汀、帕罗西汀 | 巴比妥类药物 |
| CYP3A | 咪达唑仑 (1'-羟化), 睾酮 (6β-羟化) | 酮康唑、伊曲康唑、利托那韦、葡萄柚汁 | 利福平、卡马西平、地塞米松 |
CYP3A酶的活性中心具有独特的可塑性,其能够同时容纳两个药物分子,在底物之间引发复杂的同向、异向或正向协同等非米氏动力学现象。这种特性使得预测CYP3A介导的药物相互作用变得异常复杂。例如,葡萄柚汁中的呋喃香豆素类成分能够强效抑制肠道内壁的CYP3A4,从而导致非洛地平、辛伐他汀及环孢素A等口服药物的生物利用度与血药浓度出现高达数倍的灾难性攀升。
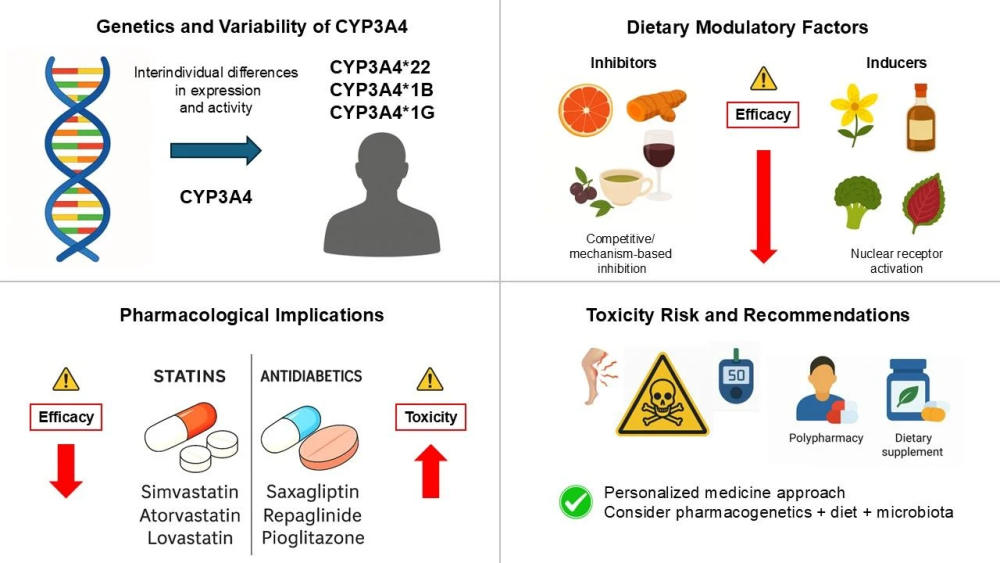
饮食对CYP3A4的调节及其对他汀类药物和抗糖尿病药物的影响(doi.org/10.3390/ph18091351)
2. 非CYP介导的氧化体系与还原、水解网络
尽管CYP450s主导了绝大部分氧化反应,但非CYP氧化酶系在特定结构药物的代谢中同样不可或缺。黄素单加氧酶(FMOs)是另一类定位于微粒体的关键酶,其主要氧化含有氮、硫、磷等杂原子的化合物。临床上具有重要意义的FMO3亚型主导了西咪替丁、氯氮平及他莫昔芬的代谢。FMOs的一大生化鉴别特征在于其高度的热不稳定性——在缺乏NADPH的体外环境中,50℃温孵仅仅1分钟即可致其完全失活,这一特性常被用于在实验中区分CYP450与FMO的代谢贡献。此外,单胺氧化酶(MAOs)和富含铁硫簇及钼结构域的钼羟化酶(如醛氧化酶)则分别在线粒体外膜和胞质中,承担着单胺类神经递质及嘌呤类药物的氧化脱氨与羟化。
在还原代谢层面,机制设计最为精妙的当属醌还原酶。该酶利用NAD(P)H作为电子供体,通过独特的“ping-pong”机制催化醌类物质的还原。在此机制中,辅酶结合后发生还原并迅速释放,随后底物才进入活性中心完成双电子还原反应。这一过程有效规避了单电子转移过程中极易引发的有害氧化还原循环及活性氧(ROS)自由基风暴,从而保护细胞免受氧化应激的毒害。
在水解代谢领域,广泛分布于内质网的羧酸酯酶(CESs)起着决定性作用。人类肝脏中高表达的CES1与肝肠均有表达的CES2,在底物口袋的立体选择性上呈现出有趣的互补。表2总结了这两种同工酶在水解前药和临床药物中的偏好性。
| 羧酸酯酶亚型 | 底物空间结构偏好性 | 典型临床底物及前药 |
|---|---|---|
| CES1 | 倾向于代谢含较小醇基或较大酰基的化合物 | 奥司他韦、可卡因甲酯、达比加群酯、依那普利、沙库必曲 |
| CES2 | 倾向于代谢含较大醇基或较小酰基的化合物 | 伊立替康、卡培他滨、可卡因苯甲酰酯、阿司匹林 |
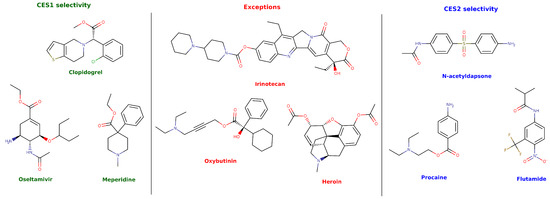
CES1 或 CES2 选择性水解的底物的分子结构,以及一般规则的例外情况。(doi.org/10.3390/jox16010011)
二、II相代谢与结合反应的解毒屏障
如果将I相代谢视为对外源分子的“分子修饰”,那么II相代谢则是真正的“解毒与排泄核心”。通过将极性亲水性内源物质(如葡萄糖醛酸、硫酸、氨基酸)共价连接至I相代谢产物或直接连接至母体药物,分子的脂溶性被彻底逆转,进而易于被跨膜转运体(如OATs、BCRP)主动泵出至尿液或胆汁中。
1. 葡萄糖醛酸化网络与UGTs家族
由尿苷二磷酸-葡萄糖醛酸转移酶(UGTs)催化的葡萄糖醛酸化结合是人体内最重要的一条II相代谢通路。UGTs主要镶嵌于内质网膜上,其催化活性位点朝向内质网腔内,必须依靠辅因子尿苷-5'-二磷酸-D-葡萄糖醛酸(UDPGA)来提供配基。
人体内的UGTs被划分为UGT1和UGT2两大主干家族。在肝脏和肠道上皮细胞中,UGT1A1、UGT1A9、UGT2B7及UGT2B15的表达最为活跃。这些同工酶的底物谱既有分工也有重叠:UGT1A1偏好催化非平面酚类及蒽醌类化合物(如抗肿瘤前药伊立替康的活性代谢物SN-38);UGT2B7则广泛参与可待因、双氯芬酸及吗啡的结合。需要强调的是,并非所有II相代谢物都毫无生物活性,吗啡经UGT2B7代谢生成的吗啡-6-葡萄糖醛酸苷,其镇痛强度不仅未减弱,反而比母体吗啡高出近百倍。而UGT1A1活性的先天性遗传缺陷,不仅会导致胆红素无法结合而引发Crigler-Najjar综合征(严重的家族性高胆红素血症),更是许多抗肿瘤药物诱发致命毒性的根源。
2. 磺酸化、乙酰化与表观解毒通路的多元延伸
硫磺转移酶(SULTs)广泛存在于各组织的细胞质中,依赖辅因子3'-磷酸腺苷-5'-磷酸硫酸盐提供的磺酸基团,对酚类及羟基固醇类底物进行硫酸化修饰。SULT1亚家族负责处理小分子酚类及雌激素,而SULT2则专门介导脱氢异雄酮等内源性胆固醇衍生物的转化。
N-乙酰基转移酶(NATs)则主要催化芳香胺及肼类药物的乙酰化。人体含有NAT1和NAT2两种亚型,其中NAT2在肠和肝中高表达,其基因序列在人类进化中保留了极高的多态性。正是NAT2的快、慢乙酰化遗传表型差异,导致了结核病患者在服用异烟肼(Isoniazid)时,遭遇肝损伤或外周神经炎毒性的概率大相径庭。
谷胱甘肽硫转移酶(GSTs)与甲基转移酶同样不可或缺。GSTs是机体应对氧化应激的急先锋,其通过催化谷胱甘肽与各类亲电子有害基团结合,保护细胞核酸与蛋白质免受不可逆的烷基化损伤。而在甲基化通路中,巯嘌呤甲基转移酶(TPMT)主导了6-巯嘌呤及硫唑嘌呤等免疫抑制剂的S-甲基化失活,TPMT基因缺失的患者在常规剂量下极易发生灾难性的骨髓造血衰竭。
三、精准药代动力学与2024-2026年CPIC权威临床指南
药物代谢酶最大的生物学特征在于其高度的遗传多态性。单核苷酸多态性(SNPs)或基因拷贝数变异(CNVs)直接导致酶活性的个体间差异,依据代谢速率,人群被区分为超快代谢型(UMs)、正常/快代谢型(EMs)、中间代谢型(IMs)和慢/弱代谢型(PMs)。近年来,临床药物基因组学实施联盟(CPIC)与FDA频繁发布和更新了基于基因型的靶向给药指南,将药代动力学彻底推向了精准医疗的深水区。
1. CYP2C19表型分化与氯吡格雷(Clopidogrel)的用药阻断
CYP2C19在亚洲人群中呈现出极高的功能缺陷频率。其中携带CYP2C19*2和CYP2C19*3(无功能等位基因)的弱代谢型(PMs)患者,在中国人和日本人群中的比例分别高达14.3%和22.5%,远超高加索人的3%。
氯吡格雷是一种经典的前体药物,必须依赖肝脏CYP2C19的代谢活化才能生成具备抑制血小板P2Y12受体功能的活性代谢产物。
氯吡格雷药物基本信息
查数据,找摩熵!图源:摩熵医药数据库
大量临床队列研究证实,在CYP2C19的IMs和PMs患者中,活性代谢物的血浆暴露量显著锐减,血小板抑制率不足,导致心血管支架置入术(PCI)后支架内血栓形成和重大心血管事件的概率呈指数级增加。
2024-2025年CPIC最新指南实现了颠覆性的更新:对于急性冠脉综合征或接受PCI的患者,一旦基因型确诊为CYP2C19中间代谢者(含一个无功能等位基因)或弱代谢者(含双无功能等位基因),强烈推荐避免使用氯吡格雷。指南要求临床医生在无禁忌症的前提下,将抗血小板方案直接替换为不依赖CYP2C19代谢的新一代药物(如普拉格雷 Prasugrel 或替格瑞洛 Ticagrelor)。此次更新将针对IMs患者的证据等级从“中等推荐”正式上调为“强烈推荐”,这标志着心血管领域的精准干预门槛被大幅收紧。
普拉格雷(左)与替格瑞洛(右)化学结构
2. CYP2D6变异与他莫昔芬(Tamoxifen)的药效挽救策略
CYP2D6虽然在肝脏的丰度仅为2%,却承担了包括三环类抗抑郁药、阿片类镇痛药等25%临床药物的代谢。该酶的基因多态性极具种族特异性:高加索人群主要表现为基因缺失或剪接缺陷诱发的完全无功能(如*3、4),而亚洲人群则广泛存在导致酶活性下降的错义突变(如10,导致第34位脯氨酸被丝氨酸替代),在中国人中发生率高达50%。
他莫昔芬作为雌激素受体阳性(ER+)乳腺癌的基础内分泌药物,其母体抗肿瘤活性极弱,需通过CYP2D6介导的连续氧化生成内昔芬(Endoxifen)方能发挥核心疗效。在CYP2D6活性低下的PMs群体中,内昔芬的生成量断崖式下跌,导致乳腺癌复发率飙升。
面对这一困境,2025版CPIC指南为他莫昔芬的处方制定了清晰的挽救路线图:针对CYP2D6弱代谢者和中间代谢者,首选策略是彻底停用他莫昔芬,改用受体拮抗机制不同且不依赖该代谢通路的芳香化酶抑制剂(Aromatase Inhibitors, AI),绝经前妇女可辅以卵巢功能抑制。若患者存在AI绝对禁忌,指南首次明确提出,在FDA批准的标签范围内,可将他莫昔芬的临床剂量翻倍至40 mg/d。药代动力学数据显示,此剂量的提升能够有效补偿代谢转化率的不足,将内昔芬浓度重新拉升至安全阈值,且不额外增加毒性风险。同时,指南严厉警告,所有服用他莫昔芬的患者必须规避与氟西汀等强/中效CYP2D6抑制剂合用,以防发生人为诱发的“表型转换”。
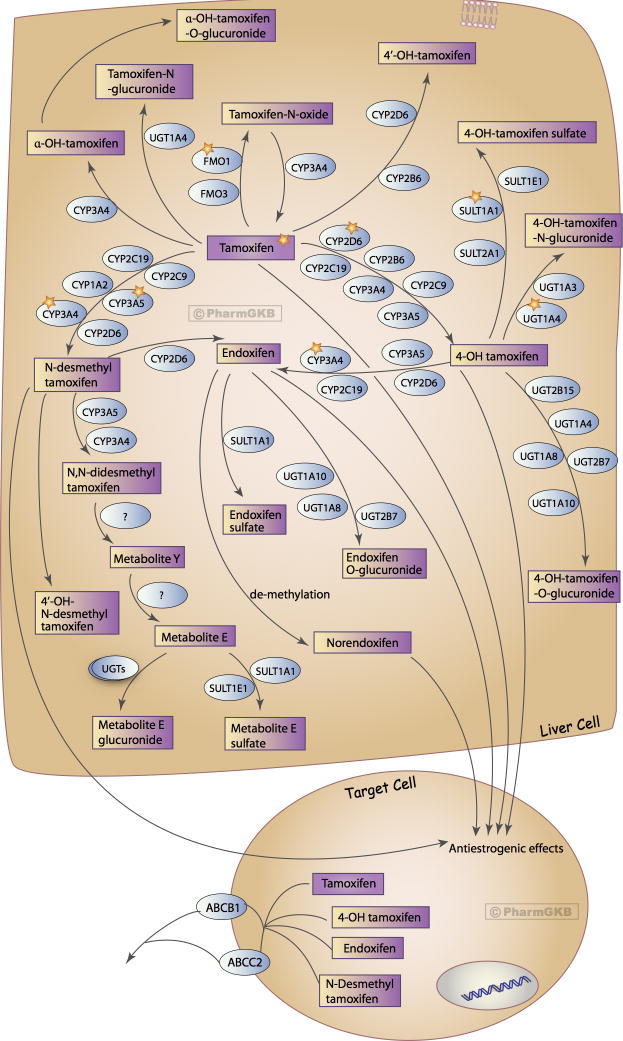
他莫昔芬通路,药代动力学(DOI:10.1097/FPC.0b013e3283656bc1)
3. UGT1A1多态性驱动的伊立替康(Irinotecan)剂量定制
如前所述,伊立替康经羧酸酯酶水解后生成的SN-38是一种极具杀伤力的拓扑异构酶I抑制剂,其细胞毒性远超母药100倍。SN-38的解毒完全依赖于UGT1A1催化的葡萄糖醛酸化。当患者携带UGT1A1功能缺陷基因时,SN-38在血液和肠道黏膜中的半衰期延长、浓度急剧蓄积,从而诱发临床难以控制的严重迟发性腹泻与致死性中性粒细胞减少症。
在欧洲与北美人群中,UGT1A1*28多态性(启动子TA重复变异)占据主导;而在亚洲人群中,UGT1A1*6突变(外显子突变)更为致命。综合2024-2026年全球顶级肿瘤学共识(如NCCN指南与DPWG指南),UGT1A1基因分型已被确立为含伊立替康化疗方案(如FOLFIRI)前必须执行的伴随诊断项目。对于被鉴定为纯合突变(*28/28或6/*6)的极高危弱代谢患者,各大指南高度统一地建议将伊立替康的初始给药剂量削减30%(即以70%的靶剂量起始)。随后的真实世界数据及前瞻性队列(如PREPARE试验)证明,这一基于药代动力学原理的预防性剂量折减,不仅将发热性中性粒细胞减少的发生率从24%断崖式压降至6.5%,且患者的最终肿瘤无进展生存期(PFS)和总生存期(OS)与接受全剂量的正常代谢者相比,未出现任何统计学劣势,完美兑现了“减毒不减效”的精准医学承诺。
四、表型转换与真实世界的多重用药博弈
尽管基于静态基因型的用药指南已日臻完善,但在2025年的临床药理学前沿,学者们越来越聚焦于一个更为复杂动态的概念——表型转换。这一现象深刻揭示了多重用药环境下,药物-药物相互作用(DDI)是如何凌驾于先天基因组之上,重塑患者的真实代谢能力的。
表型转换是指由于非遗传因素(主要是强效酶抑制剂的合并使用,或是严重新陈代谢疾病的影响),导致患者的临床表型与其基因型预测的表型出现严重错位的现象。例如,一名经基因测序确认为CYP2C19快代谢型(EMs)的消化性溃疡患者,在合并服用奥美拉唑(一种典型的CYP2C19机制性抑制剂,Ki约为3 µmol/L)后,其肝脏的CYP2C19活性被迅速且不可逆地封锁。此时,如果该患者再次服用氯吡格雷,其体内的真实代谢状态已瞬间“转换”为弱代谢型(PMs),从而面临极高的血栓风险。
为了对抗这种复杂的“药物-药物-基因相互作用(DDGI)”,现代医疗体系正大规模部署基于人工智能辅助的临床决策支持系统(CDSS)。这些系统能够实时抓取患者的基因图谱与动态电子处方池,利用系统药理学模型预测酶活性的抑制程度,从而为临床医生提供实时的剂量修正或替代药物警报,避免因盲目迷信单一基因报告而忽视表型转换带来的治疗灾难。
五、新型药物模式的非常规代谢网络挑战
随着制药工业的迭代,2024-2025年FDA批准了大量的新型药物模式(New Drug Modalities, NDMs),如抗体偶联药物(ADCs)、靶向蛋白降解嵌合体(PROTACs)以及各类多肽和寡核苷酸。这类分子因其独特的结构理化特性,对传统的以CYP450主导的I相/II相代谢评价体系提出了前所未有的挑战。
1. 抗体偶联药物(ADCs)的“潜伏代谢”与隐性DDI
ADCs由大分子单克隆抗体、可裂解连接子与小分子细胞毒性载荷三部分组成。
抗体-药物偶联物的结构
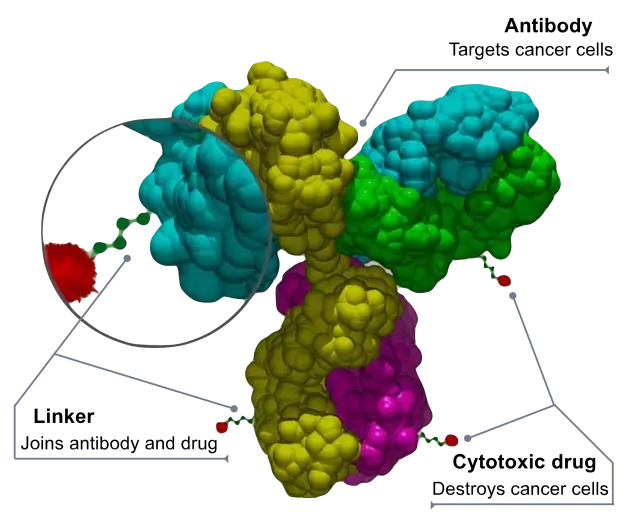
图源:Bioconjugator
虽然其在血液循环中主要依赖网状内皮系统的蛋白质水解及靶点介导的清除,但当其进入肿瘤微环境或被溶酶体降解后,释放出的高毒性小分子载荷(如MMAE、DXd、SN-38)将完全回归传统小分子的代谢与转运途径。
例如,以SN-38为载荷的ADC药物(如戈沙妥珠单抗,Sacituzumab govitecan),在靶向释放后依然会受到患者肝脏内UGT1A1多态性的制约。FDA在其说明书中明确警示,必须对携带UGT1A1*28变异的患者进行严密的毒性监测,因为这类患者在ADC释放载荷后,依然面临因葡萄糖醛酸化能力低下而诱发的不可控骨髓抑制风险。这表明,即便大分子药物的靶向性再强,其载荷的局部和全身暴露依然无法脱离II相代谢解毒酶的管控。
2. 靶向蛋白降解嵌合体(PROTACs)的超越性代谢空间
PROTACs作为一种异源双功能分子,通过一端连接靶蛋白、另一端招募E3泛素连接酶,诱导靶蛋白进入蛋白酶体降解途径。由于需要同时维持两端的亲和力与特定的连接子长度,PROTAC分子的质量通常突破800 Da,不仅彻底违背了经典的“类药五原则(Lipinski's Rule of Five)”,其在体内的代谢清除途径也大相径庭。
对于这类分子,传统的CYP450氧化通常退居次位。其PK特征更多地受到细胞膜外排转运体(如P-gp受体)、肝外组织的广泛水解以及胆汁直接排泄的影响。目前药代动力学界正在开发专门针对高分子量嵌合体的代谢稳定性模型,试图在保持其“分子胶”变构诱导活性的同时,规避肝肠首过效应的快速裂解。
六、系统生物学前沿:微生态肠肝轴与AI-PBPK建模
在微观代谢途径被逐渐探明的同时,现代药代动力学正运用宏观的系统生物学工具,将器官互作与前沿算法融入药物代谢研究的版图中。
1. 肠肝微生态轴(Gut-Liver Axis)对药物代谢的深层调控
长久以来,药物代谢被认为是单纯受宿主基因组和内环境支配的过程。然而,2025年的重磅研究深刻揭示了肠道微生物群通过“微生态-代谢物”途径对肝脏药物代谢酶表达的远程表观遗传调控。
肠道菌群不仅能够分泌特定的微生物酶直接参与口服药物在肠腔内的初级转化(如还原反应),其发酵产生的次级代谢产物——如短链脂肪酸(SCFAs)、吲哚类衍生物以及次级胆汁酸,能够穿透肠黏膜屏障进入门静脉系统。在肝细胞内部,这些菌群代谢物作为强大的内源性配体,能够与孕烷X受体(PXR)、法尼醇X受体(FXR)等关键核受体结合。受体的激活会启动下游的转录级联网络,从而系统性上调CYP3A4及UGTs等核心代谢酶的表达。这一发现解释了为何在抗生素滥用或严重肝脏疾病导致肠道菌群失调时,患者对某些治疗窗狭窄药物的清除率会发生莫名其妙的剧烈波动。因此,微生态指纹图谱正成为预测复杂药代动力学轨迹的崭新维度。
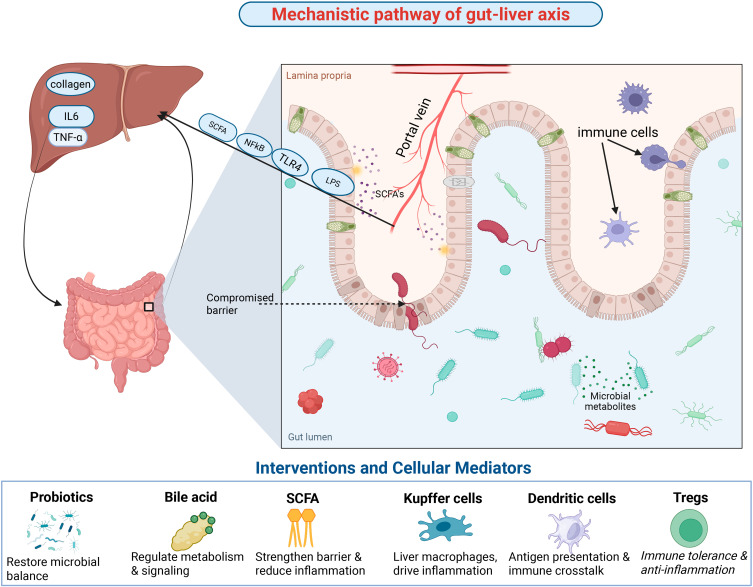
肠-肝轴的机制通路及其受干预措施和细胞介质调控的作用。肠道屏障的破坏使得微生物代谢产物和内毒素(例如脂多糖 (LPS))能够通过门静脉转运至肝脏,触发炎症信号级联反应(例如 NF-κB、TLR4),并通过胶原沉积和细胞因子释放(IL-6、TNF-α)促进纤维化。短链脂肪酸 (SCFA) 和其他微生物代谢产物可通过增强屏障完整性和调节免疫反应发挥保护作用。益生菌、胆汁酸和 SCFA 等干预措施能够恢复微生物平衡、调节代谢并减轻炎症。库普弗细胞、树突状细胞和调节性 T 细胞 (Treg) 等细胞介质进一步影响免疫串扰、抗原呈递和免疫耐受,共同作用于肝脏炎症和肠-肝稳态。(doi:10.2147/IJGM.S551494)
2. 人工智能驱动的PBPK模型(AI-PBPK)
为了在分子设计初期精准预测上述复杂的代谢波动与DDGI风险,生理药代动力学(PBPK)模型在深度学习和生成式人工智能(AI)的加持下迎来了质的飞跃。
传统的PBPK模型虽然能将生物体的血流量、组织体积与药物体外酶促动力学参数(Vmax、Km)以微分方程形式结合,但面对复杂的跨物种外推及酶竞争性抑制时常显得力不从心。新一代基于科学机器学习(SciML)的AI-PBPK架构,通过摄取海量的化学结构与多组学特征,采用图神经网络(GNNs)和Transformer模型,能够直接从分子的空间构象精准预测其在多种CYP及UGT变异体中的代谢位点(Metabolic hot-spots)和清除速率。
更具革命性的是,AI能够根据真实世界的人口学分布与遗传频率,利用物理约束扩散模型(PCDM)生成高度仿真的“虚拟患者队列”。在2024-2025年向FDA提交的新药上市申请(NDA)中,有高达26.5%的药物使用PBPK模型作为支持复杂药物相互作用和靶向人群剂量调整的关键监管证据。这标志着计算药代动力学已全面从辅助探索工具,跨越为临床试验设计和新药审批的核心支柱。
七、结论
人体对药物及外源物质的代谢清除,是一个从CYP450主导的多样化氧化,到UGTs与GSTs精密共价结合的高效分子接力过程。然而,遗传多态性及多重用药引发的“表型转换”现象,使得这一防御系统表现出极大的个体异质性。随着CPIC等国际权威指南的深度落实,靶向基因测序已成为优化特定药物(如氯吡格雷、他莫昔芬、伊立替康等)临床剂量的金标准。未来,面对ADC、PROTAC等新型药物模式的代谢挑战,深度融合AI-PBPK仿真模型、肠肝微生态轴监测的精准药代动力学体系,必将彻底重塑传统的给药模式,为患者勾勒出更为安全、高效的个体化治疗图景。
扩展阅读:
1. 2026年全球双特异性抗体核心管线深度解析:依沃西单抗OS碾压PD-1,双抗ADC强势崛起
2. BE豁免实战指南:基于BCS分类与溶出曲线f2相似因子,NMPA/FDA一致性评价降本增效策略
3. 难溶性NCE制剂决策指南:从赋能增溶技术到IVIVC体外预测的早期药物开发路径
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论