

































在现代仿制药的研发与质量一致性评价体系中,人体生物等效性试验长期以来被视为衡量仿制药与原研药是否“同工同酬”的“金标准”。然而,体内临床试验不仅周期漫长、耗资巨大,更伴随着不必要的健康人群药物暴露风险。
如何在保证药品有效性与安全性的前提下,实现研发周期的“降本增效”?人体BE试验豁免机制应运而生。通过严谨的体外药学评价或基于生物药剂学分类系统(BCS),向监管机构提供科学详实的数据,从而免除体内BE研究,这不仅是药企研发实力的体现,更是全球药政监管(包括NMPA、FDA、EMA及ICH等)趋于科学化、精细化的核心标志。
本文将从两大核心维度——基于体外试验证据的推演与基于BCS分类系统的底层逻辑,深度重构BE研究豁免的理论框架与实战策略。
一、基于体外试验证据的BE豁免策略
对于多规格口服固体制剂(涵盖常释与调释制剂),当企业已通过极其严苛的最高规格人体BE试验后,要求所有低规格再次进行体内验证显然是不符合卫生经济学的。此时,体外药学评价成为了证明不同规格间具有等效“桥接”关系的破局利器。
要跨越这道门槛,核心在于向监管证明两点:体外溶出曲线的高度重合,以及处方比例的高度相似。
1.1 溶出曲线比较:数学模型下的微观视界
溶出度不仅是质控指标,更是药物在体内释放行为的体外“模拟器”。在比较各规格制剂在不同pH介质中的溶出行为时,业界通常采用非模型依赖法与模型依赖法。
差异因子(f1)与相似因子(f2)的数理表达
在非模型依赖法中,相似因子法是最为主流且被广泛接纳的评价手段。差异因子(f1)旨在量化两条曲线在各个时间点的相对偏差度:
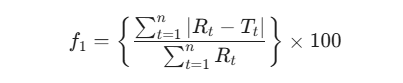
而相似因子(f2)则是一种经过对数变换的误差平方和倒数平方根指标,用于精确表征两条溶出曲线的重合度:
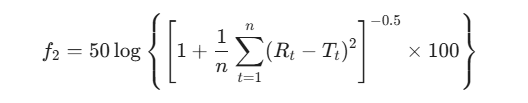
(式中,n为采样时间点数;Rt与Tt分别为参比与受试药品在t时刻的溶出率。)
【判定法则与边界条件】
- 当 f2 ≥50 且 f1 ≤15 时,两条曲线在统计学意义上被认定为相似。
- 豁免特例:若受试与参比药品在极短时间(15分钟)内溶出量即飙升至 ≥ 85%,药物在胃肠道的溶解不再是限速步骤,此时可直接判定溶出行为相似,无需再进行复杂的 f2 运算。
- 严谨性要求:变异系数(RSD)的控制是数据有效性的命门——首个取样点RSD不得逾越 20%,后续取样点则被严格限制在 10% 以内。若批内差异巨大,则需引入更为复杂的多变量置信区间(Multivariate CI)统计法。
1.2 处方比例相似性:精准到毫克的配方博弈
仅有溶出相似不足以服众,制剂处方的“骨架”必须保持一致。所谓“比例相似”,在药政语境下被严格界定为以下两种路径:
路径一:绝对的等比例缩放
这是最理想的状态。不同规格间,活性成分(API)与所有非活性辅料的质量占比分毫不差。例如,某80mg规格与已通过BE的160mg规格,其辅料比例完全锁定,仅整体片重减半。此类情形只要溶出曲线达标,豁免几乎是水到渠成。
路径二:非等比例变化的边界
在实际工业放大或处方调整中,往往难以做到绝对等比例。此时,辅料用量的变动幅度必须被限制在极小的允差范围内(通常参考SUPAC指南中的II类变更范围,如一般填充剂变化限度为 ±10%)。
【深度解析:高活性药物的处方博弈】
对于高活性、低剂量药物(API占比 < 5%),微小的辅料变动可能被放大。此时的铁律是:不同规格制剂的总重波动必须死死卡在 ±10% 之内。规格的调整只能通过微调API本身和一至多种无特定释药控制功能的辅料来实现,且所有规格必须使用完全相同种类的辅料库。
典型案例逻辑解构:以某缓释制剂为例,若200mg为BE基准规格,拟豁免的150mg规格中非释药控制性辅料的比例变化幅度为3.5%(< 10% 的II类变更红线),则具备豁免资格。反之,若某一规格(如50mg或400mg)的辅料波动幅度达到15%或19%以上,严重突破了比例相似的红线,且非高活性药物,其豁免申请将被监管机构无情否决。
二、生物药剂学分类系统(BCS)的底层逻辑
如果说基于体外试验证据的豁免是战术上的桥接,那么基于BCS分类系统的豁免则是对药物在人体内吸收机制的降维打击与战略重构。
1995年,Amidon教授团队首次提出BCS系统,犹如在混沌的药物吸收理论中建立了一套坐标系。该系统摒弃了繁杂的表象,直击决定口服固体常释制剂吸收速率与程度的三大命门:药物溶解性、肠道渗透性以及制剂溶出度。
2.1 吸收动力学的物理学溯源:Fick第一定律
BCS理论的根基并非无源之水,它深植于经典的物理扩散定律——Fick第一定律。药物在胃肠道黏膜的吸收本质上是一个跨膜扩散过程。单位时间内的药物扩散通量(J)与浓度梯度成正比:
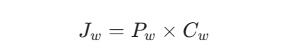
在此基础上,药物在肠道表面(面积为 A)的总吸收质量 M(t) 可积分表达为:
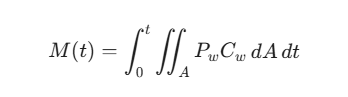
(式中,Pw 为胃肠黏膜有效渗透系数;Cw 为黏膜表面的微环境药物浓度。)
这一组数学物理方程揭示了一个极其重要的药政逻辑:如果两种制剂(仿制与原研)具有相同的有效成分,且在胃肠道环境中具有完全重合的溶出曲线(保证了完全一致的 Cw 时间序列),且处方中没有任何干扰渗透系数 Pw 的“作弊”辅料,那么这两种制剂在人体内的吸收速率和程度必然是高度一致的——这即是BCS豁免机制在微观层面的绝对法理依据。
2.2 BCS的四象限法则与全球监管共识
依据溶解性与渗透性的高低,BCS将所有API无情地划分为四大阵营:
- BCS I类:高溶解,高渗透。
- BCS II类:低溶解,高渗透。
- BCS III类:高溶解,低渗透。
- BCS IV类:低溶解,低渗透。
BCS分类
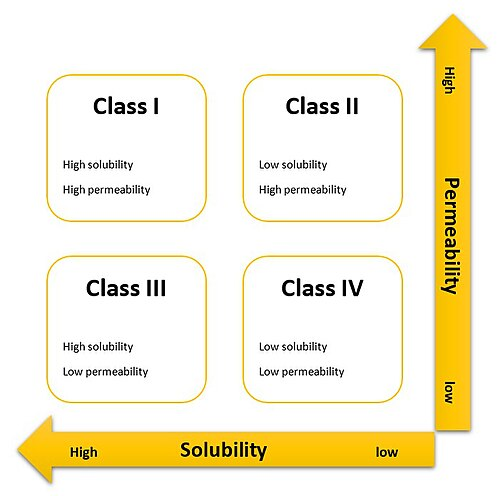
【监管共识矩阵】各大监管机构(NMPA, FDA, EMA, WHO, ICH)在经历了多年的数据积累与理论探讨后,达成了一项关键共识:BCS I类与部分BCS III类常释固体制剂,是赋予BE豁免路权的绝对主力梯队。
三、如何界定“高溶解”与“高渗透”?
在研发实战中,证明一个药物属于I类或III类,需要极其缜密的体外测定数据支撑。这里的每一个定义都伴随着严格的定量边界。
3.1 溶解性:250mL的生理学隐喻
药政意义上的“高溶解性”,并非指药物在烧杯中的绝对溶解度,而是基于人体生理特征的相对考量。
判定标准:在37∘C±1∘C环境下,于pH 1.0~6.8的生理范围内,药物的最大单次给药剂量能够完全溶解于 250mL 缓冲液中。
- 为什么是250mL? 这一数字高度还原了标准BE试验中受试者用一杯水(250mL)送服药物的真实场景。
- 操作严谨性:需涵盖API的pKa两侧节点(pKa+1, pKa-1)及极值点,每个pH点至少平行测定3次,且必须使用经方法学验证(区分降解产物)的含量测定法。
3.2 渗透性:跨越肠壁屏障的 ≥85% 红线
渗透性分类直接映射了药物在人体内的吸收程度。当一个药物被证实其体内吸收程度(不等于系统生物利用度,需排除首过效应)达到或超过 85% 时,即可被加冕为“高渗透性”药物。
测定渗透性是一项技术密集型工作,业界通常采纳以下梯队方法:
(1)人体绝对BA与质量平衡研究(金标准):放射性同位素标记下,若原形药物及代谢物在尿液中回收率 ≥85%,或绝对生物利用度 ≥85%,且证实胃肠道内稳定性良好(非降解损耗),即可一锤定音。
(2)体内/原位肠道灌注模型:直接测定膜转移速率。
(3)体外细胞模型(如Caco-2单克隆细胞系):适用于被动转运机制的药物。
【警惕外排蛋白(Efflux Transporters)的陷阱】在渗透性评估中,最狡猾的干扰项是肠道外排转运蛋白(如P-gp、BCRP)。某些药物在体外看似渗透性极佳,但由于是P-gp的底物,在人体内会被无情地“泵”回肠腔。因此,在依赖体外或动物模型数据时,必须提供确凿证据证明该药物的吸收机制为被动转运:例如,证明其在临床剂量下呈现药代动力学线性关系,或者体外双向转运实验中外排比(外排率)< 2。同时,必须引入多达20种已知的高/中/低渗透性模型药物(如普萘洛尔、雷尼替丁、甘露醇)及零渗透性内标(如PEG-4000)作为体系验证的锚点。
3.3 溶出度评价:体外与体内的动态映射
采用篮法(100 rpm)或桨法(50/75 rpm),在900mL(或500mL)的pH 1.2/4.5/6.8介质中,必须取得12个单剂量药品的溶出曲线。
- BCS I类豁免:要求试验与参比制剂均具备“快速溶出”特性(30分钟内溶出 ≥85%)。
- BCS III类豁免:由于吸收受限,对溶出的要求被拔高至“非常快速溶出”(15分钟内溶出 ≥85%)。
四、辅料对体内吸收的深度干扰
在撰写一致性评价报告时,许多研发人员容易陷入一个误区:只要原料药是BCS I或III类,豁免就稳操胜券。事实上,辅料才是决定豁免成败的隐藏Boss。
辅料早已不再是单纯的“惰性”赋形剂,它们具有复杂的生物药剂学活性,能够通过改变胃肠动力、渗透压、微环境pH甚至直接干预转运蛋白,重塑药物的体内命运。
4.1 BCS I类 vs. BCS III类的辅料豁免门槛
- 对于BCS I类药物:门槛相对宽容。只要使用的是NMPA已批准的常释制剂常用辅料,且用量符合其功能预期(如常规润滑剂),通常不会颠覆其本就强势的吸收特性。但若大量使用表面活性剂(吐温80)或高渗透压甜味剂,仍需警惕。
- 对于BCS III类药物:门槛极其严苛。由于低渗透性是其先天软肋,辅料的任何风吹草动都可能引发蝴蝶效应。因此,受试药品的辅料种类必须与原研参比药品完全保持一致,且用量必须极其相似(累计变化量严控在总重的10%以内)。
4.2 重点辅料的机制拆解
结合FDA Inactive Ingredient Database(IIG)的逻辑,我们必须对以下高能辅料保持十二分的敏锐度:
- 糖醇类(甘露醇、山梨醇):典型的渗透压引擎。它们难以被肠道吸收,会在小肠内引发水通量的变化(跨膜拖曳效应)。文献表明,随着山梨醇用量的激增,BCS II类药物雷尼替丁的生物利用度会发生断崖式下跌(降幅高达45.5%)。
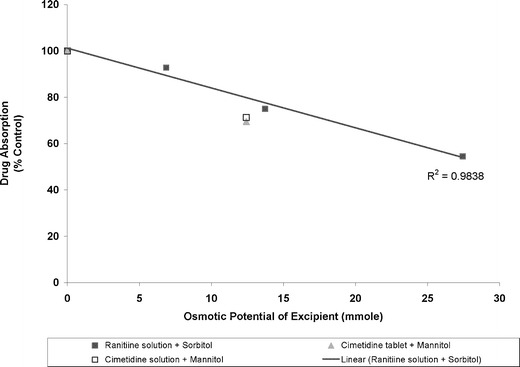
山梨醇/甘露醇渗透势与雷尼替丁/西咪替丁吸收的关系(与对照组比较)(doi:10.1208/s12248-013-9509-z)
- 聚乙二醇(PEG):机制复杂的“多面手”。它不仅产生渗透稀释作用,还能直接调节膜流动性。更致命的是,特定分子量的PEG能对P-gp等外排蛋白产生浓度依赖性的抑制,彻底打乱肠道通透性的固有节奏。
- 表面活性剂(SDS、聚山梨酯80):渗透促进剂。4mg的十二烷基硫酸钠(SDS)甚至能将某些低渗药物的BA暴力拉升5倍之多;而聚山梨酯80作为P-gp的强效抑制剂,能显著提高环孢素等底物药物的表观渗透系数。
- 高分子聚合物(泊洛沙姆、卡波姆):泊洛沙姆可通过减缓肠道蠕动变相增加吸收窗口期;而卡波姆作为黏合剂,甚至能直接对特定转运体(如Pept1)的高亲和力底物(如福辛普利)产生空肠吸收的靶向促进作用。
因此,对于仿制药研发而言,逆向工程不仅要破解原研的API晶型,更要对原研辅料的种类与微观用量进行极其精准的破译与镜像复制。
4.3 豁免的绝对禁区
任何规则都有红线。以下领域绝对不适用BCS豁免机制:
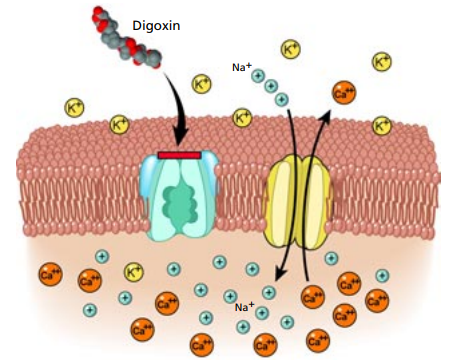
(1)治疗窗狭窄药物(NTI):如地高辛、华法林。微小的浓度波动即可引发毒性或失效,绝无宽容空间。
地高辛的作用机制
(2)非胃肠道吸收制剂:BCS理论构建于小肠生理学之上,对于舌下片、口腔崩解片等通过口腔黏膜吸收的系统,因渗透环境截然不同,该理论直接失效。
(3)存在PK干扰的复方制剂(FDCs):若复方中混入了非I/III类药物,或组分间存在代谢酶竞争等药物相互作用,则必须通过真实体内试验进行确证。
结语
人体生物等效性研究豁免,绝非药政监管的“放水”,而是现代制药科学从“黑箱测试”向“机理预测”演进的伟大里程碑。无论是基于严谨体外溶出曲线及处方比例桥接的规格豁免,还是基于Fick扩散定律的BCS系统分类豁免,其底层逻辑都指向了同一个终极命题:用最高维度的数据解析,换取最大程度的确定性。
对于立志于拓展全球市场、深耕重磅仿制药管线的医药企业而言,深刻理解这套体系,熟练运用溶解性评估、Caco-2细胞模型以及对各类高能辅料的机制干预,将成为决胜一致性评价、抢占首仿时间窗口的核心商业武器。在药学的世界里,真正的高手,往往能在试管与计算之间,提前锁定体内吸收的胜局。
扩展阅读:
1. 难溶性NCE制剂决策指南:从赋能增溶技术到IVIVC体外预测的早期药物开发路径
2. 二甲双胍降糖效果因人而异?OATPs/OCTs/MATEs五大SLC转运体家族揭秘个体化用药真相
3. 2026年值得关注/追踪的抗体偶联药物:T-DXd、Dato-DXd、双抗ADC谁主沉浮?
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论