


































做药代研究的人,大概都见过这样的场景:一个化合物在肝微粒体中表现稳定,换到肝细胞后却明显加快消失;也可能恰好相反——微粒体中清除很快,肝细胞里却像一个“慢代谢化合物”。更棘手的是,体外体系看起来都很稳定,动物体内的血浆浓度却下降得远快于预期。
对 PROTAC 而言,这些结果并不必然意味着实验失败。更常见的情况是,不同模型观察到的是同一个分子的不同处置环节。肝微粒体主要呈现微粒体酶环境;完整肝细胞同时引入细胞膜、胞质酶、部分转运过程和更完整的辅因子体系;肝胞质和 Liver S9 有助于发现微粒体遗漏的胞质代谢;血浆与全血则用于判断循环中是否存在化学降解、酶促水解、血细胞分配或细胞内代谢。
因此,PROTAC 代谢研究的核心并不是寻找一个“最权威”的单一模型,而是先明确当前要回答的问题,再判断所选模型是否具备回答这个问题的能力。
一、为什么 PROTAC 的代谢研究比普通小分子更容易“绕路”
典型 PROTAC 由 POI 配体、E3 连接酶招募配体和 Linker 三部分组成,常处于 beyond-rule-of-five 化学空间。它们往往分子量较大、可旋转键多、氢键供受体多,并同时面临溶解度、膜通透、蛋白结合和非特异性吸附等问题。完整分子也不是两个已知配体的简单相加,而是一个新的化学实体:连接后构象、极性暴露、分子内氢键和酶结合方式都可能改变。
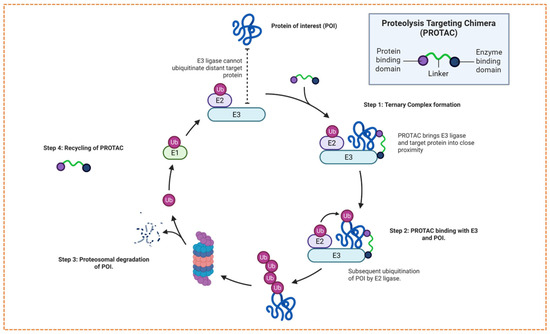
PROTAC的设计与作用机制概述,利用细胞蛋白酶体系统及PROTAC介导的蛋白质降解
因此,单独 POI 配体稳定、E3 配体稳定,并不能推出完整 PROTAC 同样稳定。完整分子的主要代谢软点可能出现在 Linker、连接位点、配体暴露面,或由连接后形成的新构象所暴露的位置。反过来,单独配体中明显的软点也可能在完整 PROTAC 中被折叠或空间遮挡。
这意味着第一条原则应当是:完整 PROTAC 必须作为主要研究对象;单独配体、Linker 模型化合物和断裂片段的数据可以辅助解释,但不能替代对完整分子的评价。
二、先定义问题,再选择模型
固定地按“微粒体—肝细胞—血浆稳定性”执行一套套餐,能够提供基础信息,却未必能解释异常结果。更有效的方法,是把问题拆成以下几个层级。
1. 是否主要由微粒体酶介导代谢
如果目标是快速比较同系列化合物的氧化代谢倾向,肝微粒体通常是高效的起点。加入 NADPH 后,体系主要覆盖 CYP 和 FMO 等微粒体氧化酶;在专门加入 UDPGA 等辅因子并优化膜通透条件时,也可研究部分 UGT 反应。因此,把肝微粒体简单等同于“只看 CYP”并不严谨,但在常规 NADPH 稳定性实验中,CYP 相关氧化通常是主要信息来源。
微粒体体系结构简单、通量高、重复性较好,适合发现羟基化、N-去烷基化、O-去烷基化等反应,也便于开展重组酶、化学抑制和代谢表型研究。其局限同样明确:它缺少完整细胞膜、胞质酶、主动摄取和外排过程,对某些结合代谢和细胞内处置覆盖不足。
所以,“微粒体稳定”只能说明在该孵育条件下,微粒体相关代谢较慢;它不能直接等同于肝脏清除慢,更不能等同于人体总清除慢。
2. 是否需要观察完整肝细胞中的综合处置
肝细胞保留细胞膜、胞质和更完整的代谢酶体系,能够同时反映 Phase I、部分 Phase II、细胞摄取和外排等过程,因此通常比单一亚细胞组分更接近肝内真实环境。
但“更接近生理”不等于“自动代表完整肝脏”。悬浮肝细胞的细胞极性、胆小管结构和部分转运功能有限;冷冻复苏、供体差异、细胞活性和孵育时间也会影响结果。对 PROTAC 来说,观测到的表观内在清除率还可能受到膜通透、主动摄取、外排、细胞内结合及非特异性吸附共同控制。
因此,同一个化合物在微粒体中代谢快、在肝细胞中却显得稳定时,不能立刻把它解释为“完整细胞更稳定”。需要继续区分:
- 是否因被动通透低或外排强,导致细胞内游离浓度不足;
- 是否存在高细胞或培养基结合,使真正可被酶利用的浓度降低;
- 是否存在微粒体与肝细胞酶活差异;
- 是否因溶解度、吸附或缓慢平衡造成表观母体损失或假性稳定。
对低清除化合物,短时悬浮肝细胞实验可能触及灵敏度下限。此时可考虑延长孵育、hepatocyte relay 或长期培养体系,但必须同时验证细胞功能、回收率和化合物稳定性。
3. 是否存在胞质酶或肽酶参与
肝胞质和 Liver S9 的价值,在于发现常规微粒体容易遗漏的代谢通路。Liver S9 同时包含微粒体和胞质成分,但酶活被稀释,且不同反应需要匹配的辅因子;如果怀疑某一具体胞质酶,直接使用肝胞质并开展抑制或酶学验证往往更清晰。
醛氧化酶(AOX1)主要识别某些电子缺陷的含氮杂环,具有明显的结构依赖性和物种差异。对含吡啶并嘧啶、喹唑啉、嘌呤样或其他潜在 AO 易感杂环的 PROTAC,可在人肝胞质、S9 或肝细胞中结合选择性抑制和代谢物结构进行判断。需要强调的是,不能因为 PROTAC 分子大或含 VHL 配体,就默认 AOX1 一定参与。
此外,部分 VHL 型 PROTAC 已观察到 VHL 配体区域的特征性裂解,研究提示肝内和肝外脯氨酰内肽酶可能是重要参与者。这个例子说明,PROTAC 的非典型代谢并不限于 AOX1:当代谢物表现为特征性片段丢失时,应从片段结构反推可能的肽酶、酰胺酶或其他水解酶,而不是只在 CYP 和 AO 两类酶中寻找答案。[3]
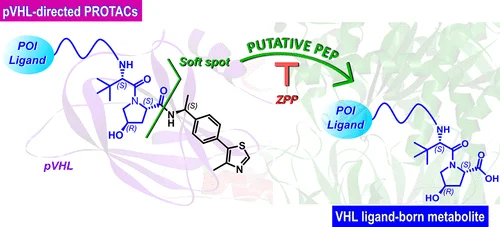
4. 分子是否会在循环系统中降解或发生血细胞处置
血浆与全血稳定性适合研究循环中的化学降解、酶促水解和血细胞相关处置。需要重点关注的结构包括酯、碳酸酯、部分氨基甲酸酯、酰亚胺、内酰胺,以及具有明显肽样识别特征的酰胺连接。某些 CRBN 配体本身也可能存在水解或构型稳定性问题,且连接位点会显著影响稳定性。[4]
普通酰胺和磺酰胺通常不能仅凭键型就归为“易水解”。它们是否不稳定,取决于电子效应、邻近基团、构象暴露和是否能够被特定酶识别。
血浆稳定而全血中母体下降,并不只意味着血细胞酶参与。还可能是化合物快速进入红细胞、与细胞成分强结合,或在分离血浆时造成回收偏差。理想的设计应同时考察:完整全血中的总母体回收、分离血浆中的浓度、血细胞/血浆分配比、代谢物形成和样品处理稳定性。
新鲜样品通常更有利于保留接近生理状态的酶活和细胞功能,但不能笼统地说新鲜全血一定优于冻存血浆。应根据问题选择匹配基质,并控制物种、抗凝剂、温度、冻融次数、孵育时间和样品采集流程。
三、需要重点关注的代谢力量
1. CYP3A 常常重要,但不能预设为唯一主力
许多 PROTAC 具有较高脂溶性、较大的疏水表面和柔性 Linker,因此 CYP3A 家族经常参与其氧化代谢,Linker 烷基链、PEG 样链段及两端配体均可能发生羟基化或去烷基化。但“经 CYP 代谢”只是起点,不能据此默认 CYP3A4 是唯一或绝对主导酶。
更有价值的问题包括:代谢发生在哪个结构模块;是单一步骤还是连续多步氧化;不同 CYP 亚型的相对贡献是多少;抑制某个软点后是否发生代谢转移;结构修改是否同时损害三元复合物形成、降解活性、通透性或溶解度。
2. 胞质酶与肽酶是微粒体之外的重要盲区
AOX1、黄嘌呤氧化酶、羰基还原酶、醛酮还原酶以及多类肽酶、酰胺酶,都可能在特定结构上发挥作用。是否需要前置这些研究,应由结构警示和模型差异驱动,而不是对所有 PROTAC 无差别加做。
判断某一通路时,最好形成“模型差异—选择性抑制—代谢物结构—重组酶或酶源验证”的证据链。单凭加入一个抑制剂后母体消失变慢,通常只能说明该通路可能参与,还要排除抑制剂非特异作用和体系扰动。
3. 水解酶可能改变母体,也可能改写 PK/PD
如果 Linker 或 E3 配体区域发生断裂,产生的 POI 片段、E3 配体片段或半完整降解剂,通常具有与母体完全不同的分子量、极性、蛋白结合、组织分布和清除行为。
这些片段并非必然保留药理作用。只有当其体内暴露足够、仍具有可观的靶标或 E3 结合能力,并能到达相应组织时,才可能与母体竞争、产生独立抑制、影响 E3 相关蛋白相互作用,或带来额外的脱靶风险。因此,片段风险应以“结构—暴露—活性”三者共同评价,而不是只凭结构推测。
四、模型之间不一致,往往是最有价值的信息
微粒体快、肝细胞慢
这类结果可能由低被动通透、强外排、高细胞结合或培养基结合导致,使化合物难以在细胞内形成足够游离浓度;也可能源于微粒体与肝细胞中相关 CYP 活性的差异。下一步应优先检查细胞摄取或胞内暴露、孵育体系 fu,inc、回收率、溶解度和吸附,而不是直接把肝细胞结果解释为高稳定性。
微粒体慢、肝细胞快
这可能提示胞质酶、结合代谢、主动摄取或多步串联代谢参与。可进一步比较 S9、胞质和肝细胞,加入匹配辅因子和选择性抑制剂,并通过 MetID 判断新增通路。
血浆稳定、全血中母体下降
除了血细胞内或细胞表面酶促代谢,还要考虑红细胞分配、细胞摄取和样品分离造成的表观损失。若全血总回收良好、但血浆母体下降且血细胞中浓度升高,更符合分配而非降解;若同时出现特异代谢物,则支持血细胞相关代谢。
体外稳定、体内血浆浓度仍快速下降
首先要区分“血浆浓度下降”和“真实消除清除增加”。给药后的早期快速下降可能主要来自组织分布,而不一定代表代谢或排泄快;应结合静脉给药 PK、分布容积、终末相、质量平衡和组织浓度判断。
若确认总清除确实较高,再考虑肝外代谢、胆汁或肾排泄、主动摄取转运、血液中降解、肠肝循环缺失,以及体外体系未覆盖的酶。对某些 VHL 型 PROTAC,已有研究显示血浆暴露很低但组织摄取和滞留显著,说明单看血浆 PK 可能低估靶组织暴露和持续药效。[6]
五、高蛋白结合、非特异性吸附和低溶解度必须一起看
PROTAC 常见高血浆蛋白结合和高孵育体系结合。对于代谢清除,真正与酶或转运体发生作用的是相应环境中的游离化合物,因此需要区分血浆游离分数 fu,p 和孵育体系游离分数 fu,inc。二者不能互相替代。
尤其要警惕直接套用针对普通小分子建立的 fu,inc 预测公式。2025 年一项 PROTAC 数据研究发现,传统公式显著高估部分化合物在肝细胞孵育中的游离比例;改用实验测定的 fu,inc 后,IVIVE 的系统性低估有所改善,但仍未完全解决。[1]
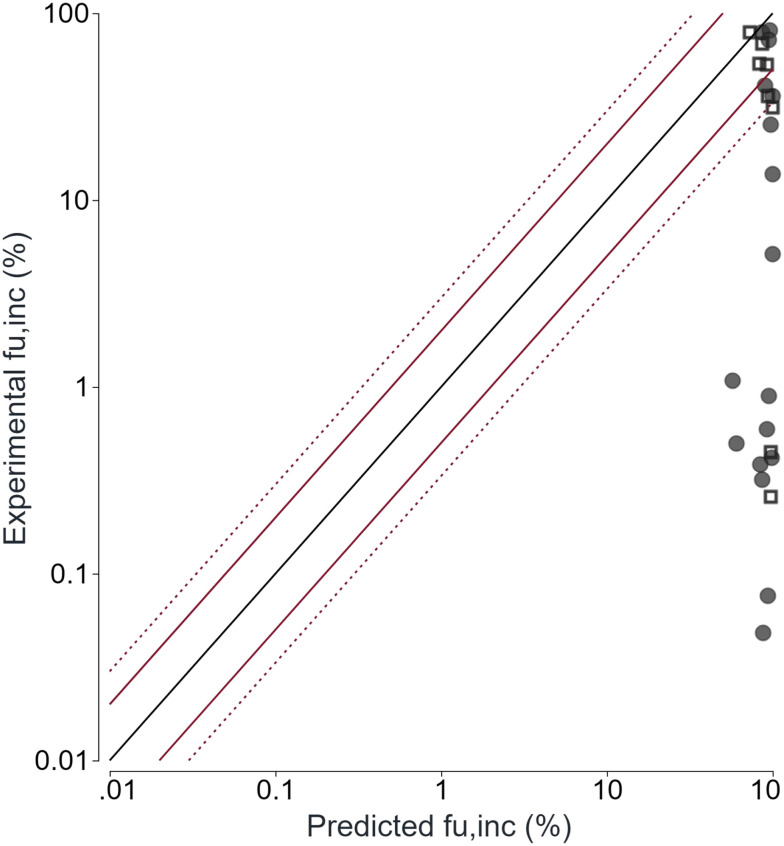
蛋白结合还必须与非特异性吸附和沉淀区分。化合物可能黏附在塑料管、培养板、透析膜和移液耗材表面,也可能因工作浓度高于动力学溶解度而析出。此时母体下降并不等于代谢,低接收侧浓度也不等于低通透。
因此,任何关键稳定性或通透性实验都应把总回收率、空白基质稳定性、无酶对照、容器吸附和浓度依赖性作为结果解释的一部分。近期对临床阶段 PROTAC 的系统测试也表明,低溶解度、高非特异性结合和传统 ADME 方法的可转译性不足,是这一分子类别的普遍实验挑战。[2]
六、找到代谢软点以后,不是简单把它“堵死”
PROTAC 的代谢软点可能位于 POI 配体、E3 配体、Linker 主链、连接位点,或特定可水解结构。常见优化方式包括调整连接向量、改变电子性质、引入适度位阻、局部氟代、环化或刚性化 Linker,以及以更稳定的生物电子等排体替换易裂解基团。
但每一次稳定化都可能改变整个分子的构象集合。增加位阻可能提高代谢稳定性,却破坏三元复合物几何;把柔性 Linker 刚性化可能减少氧化位点,也可能降低蛋白表面适配;增加脂溶性有时改善膜进入,却可能进一步降低溶解度、提高非特异性结合和组织蓄积。
更合理的目标不是获得“永不代谢”的分子,而是在降解活性、选择性、细胞内暴露、溶解度和体内清除之间取得可预测的平衡。PROTAC 还可能通过环境依赖的折叠和极性遮蔽获得膜通透,因此设计时应关注暴露极性和构象可塑性,而不能只看二维分子量或总极性表面积。[5]
七、代谢物鉴定应贯穿发现、临床前和临床阶段
发现阶段:回答“主要从哪里坏掉”
早期 MetID 的重点不是穷尽所有低丰度峰,而是尽快定位主通路和可操作软点。建议同时比较微粒体、肝细胞、S9/胞质和血液基质中的主要代谢物,特别关注 Linker 断裂、E3 配体水解、连续氧化及两端配体相关片段。
临床前阶段:回答“动物是否覆盖人体通路”
应比较人和毒理候选物种的体外代谢谱与速率,并结合动物体内血浆、尿液、粪便、胆汁和组织样品判断体内相关性。物种比较不应只看是否“出现同一个峰”,还要比较相对暴露、形成速率和后续清除。
临床阶段:回答“人体真正暴露了什么”
进入人体后,需要确认母体主要清除路径、人体特异或高暴露代谢物,以及关键断裂片段是否具有持续暴露和药理活性。对具有显著组织滞留的 PROTAC,血浆中的代谢物谱也可能无法完整代表组织内过程,必要时需结合可获得的组织、生物标志物或成像数据进行解释。
对于已经进入临床或接近临床阶段的同靶点项目,还可以通过摩熵医药数据库关联查看全球研发进度、临床试验设计及公开药代信息,重点比较给药途径、暴露水平、给药频率和已披露的安全性信号。
靶向ALK的全球新药
查数据,找摩熵!图源:摩熵医药数据库
这样的横向对照有助于识别本项目的异常究竟来自单个分子的结构问题,还是该靶点、E3 配体或分子类别的共性挑战;但任何跨项目比较都应同步核对剂量、物种、检测方法和样本基质。
八、PROTAC 代谢物的半定量尤其容易产生偏差
高分辨质谱能够发现代谢物,但峰面积不等于摩尔量。完整 PROTAC 及其代谢物可能形成单电荷、多电荷、不同加合离子和源内裂解离子;Linker 断裂后,片段的离子化效率可能与母体相差数个数量级。只积分一个离子,或者直接用母体响应因子估算所有代谢物,都可能严重误判相对丰度。
较稳妥的做法包括:确认主要电荷态和加合离子;通过源参数变化识别源内裂解;使用同位素或结构相近内标;对关键代谢物合成标准品;必要时采用放射性质量平衡或定量 NMR 支持。UV 也并非天然“等响应”,因为不同片段保留的发色团和摩尔吸光系数可能不同。
因此,无标准品的 LC-HRMS 峰面积更适合用于“排序和发现”,而不是声称绝对代谢物比例。真正影响安全性或物种覆盖判断的代谢物,应尽可能升级为有标准品的定量。
九、IVIVE 不是把体外数字乘一个系数
IVIVE 的基本逻辑,是把体外测得的内在清除按肝细胞数或微粒体蛋白量放大到全肝,再结合肝血流、血液/血浆分配和游离分数,通过 well-stirred 等模型预测肝清除。对 PROTAC 而言,任何一个输入项偏差都可能被放大。
至少需要关注以下因素:
- 体外模型是否覆盖主要清除通路;
- fu,p、fu,inc 和血液/血浆比是否为可靠的实验值;
- 肝细胞内暴露是否受通透、主动摄取或外排限制;
- 是否存在显著非特异性吸附、沉淀或低回收;
- 体内是否有肝外代谢、胆汁排泄、肾排泄或血液降解;
- 表观血浆下降是否混入快速组织分布。
关于“多数 PROTAC 能否预测在三倍范围内”,目前不能给出脱离数据集的统一答案。较早的一组 27 个内部化合物中,16 个(59%)由小鼠肝细胞预测的内在清除位于三倍范围内,但该数据集偏向高内在清除。另一项 2025 年研究对 25 个 PROTAC 使用标准小分子方法时,仅 6 个(24%)位于三倍范围内;改用实验测定的 fu,inc 后提高到 10 个(40%),仍说明该方法尚不能被视为普遍可靠。[1]
因此,更稳妥的策略是:在同一化学系列内,用实测 fu,inc、fu,p、血液/血浆比和体内静脉 PK 建立本系列的经验关系;随着化合物增加持续校正偏差;对异常点回到通路、回收和组织分布层面解释,而不是用一个通用校正因子覆盖所有结构。
十、血浆PK不是 PROTAC 药效的唯一暴露指标
PROTAC 的药理是事件驱动的:只要在细胞内形成足够的有效三元复合物并触发泛素化,母体浓度下降后,蛋白恢复还需要重新合成。因此,血浆浓度、组织内母体、细胞内游离浓度和蛋白降解效应之间可能存在明显时间滞后。
泛素化的类型
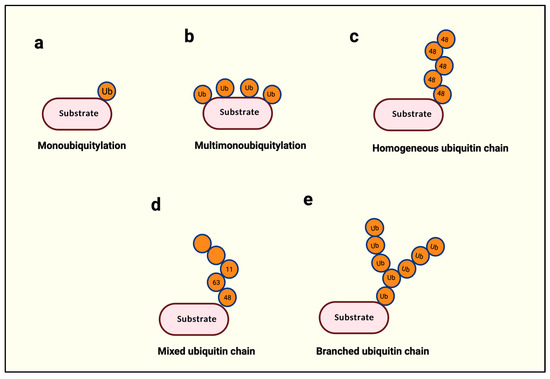
某些快速从循环中消失的 VHL 型 PROTAC 仍可在特定组织和肿瘤中摄取、滞留,并维持较长时间的蛋白降解。这类现象提示,开发阶段应把血浆 PK 与靶组织暴露、细胞内累积、降解深度和恢复动力学联合分析,而不能仅凭血浆半衰期判断给药频率或药效持续时间。[6]
当然,组织滞留也不是天然优势。若分布到非靶组织、难以清除或形成高暴露活性片段,同样可能带来安全性风险。因此,组织分布需要与靶点表达、E3 表达、药效窗口和毒理信号共同解释。
十一、把代谢研究变成药物化学的反馈系统
一套更具机制性的 PROTAC 代谢研究流程,可以按以下顺序推进:
- 先做基础质量控制:溶解度、化学稳定性、容器吸附、总回收率和 LC-MS 方法适用性;
- 用肝微粒体和肝细胞进行初筛,同时测定或评估 fu,inc,识别模型间差异;
- 根据结构警示和差异,选择 S9、肝胞质、特定辅因子、抑制剂、重组酶或血液基质;
- 通过 HRMS MetID 定位主通路和结构软点,并确认母体下降是否伴随代谢物形成;
- 补充 fu,p、血液/血浆比、全血稳定性和关键转运/摄取信息;
- 开展静脉 PK 和必要的组织暴露研究,区分分布与消除,并建立本系列 IVIVE;
- 把“软点位置、限制步骤和暴露后果”反馈给药物化学,而不是只返回一个半衰期数字;
- 新一轮化合物进入同一闭环,持续验证结构修改是否真正改善了整体 DMPK,而不是只改善单项指标。
这套流程的价值,不是给每个化合物盖上“稳定”或“不稳定”的标签,而是回答三个连续问题:它在哪里消失;为什么在这个模型中消失;这种消失是否真的限制体内靶组织暴露和药效。
结语:选对模型,更要读懂模型
PROTAC 代谢研究的困难,不只来自分子量大和结构复杂,更来自代谢、膜转运、结合、吸附、组织分布和事件驱动药理之间的高度耦合。任何单一模型都难以给出全部答案。
肝微粒体适合高通量观察微粒体酶相关代谢;肝细胞提供更完整但仍有限的细胞环境;S9 和肝胞质帮助发现胞质通路;血浆与全血用于判断循环稳定性、血细胞分配和血液相关代谢;组织暴露和 PK/PD 则用于确认血浆数据能否代表真正的作用部位。
当这些模型围绕明确问题进行组合,数据差异就不再只是麻烦,而会成为机制线索。对 PROTAC 来说,最好的代谢研究方案不是实验项目最多,而是能以最少但足够的模型,逐层识别限制步骤,并把结果转化为下一轮可执行的分子设计。
参考文献:
[1] Maurer CK, Fang Z, Schindler C, et al. In vitro and in vivo ADME of heterobifunctional degraders: a tailored approach to optimize DMPK properties of PROTACs. RSC Med Chem. 2025;16(4):1746-1757. doi:10.1039/D4MD00854E.
[2] Srivastava A, Pike A, Swedrowska M, Nash S, Grime K. In Vitro ADME Profiling of PROTACs: Successes, Challenges, and Lessons Learned from Analysis of Clinical PROTACs from a Diverse Physicochemical Space. J Med Chem. 2025;68(9):9584-9593. doi:10.1021/acs.jmedchem.5c00358.
[3] Castellani B, Eleuteri M, Di Bona S, et al. VHL-Modified PROteolysis TArgeting Chimeras (PROTACs) as a Strategy to Evade Metabolic Degradation in In Vitro Applications. J Med Chem. 2023;66(18):13148-13171. doi:10.1021/acs.jmedchem.3c01144.
[4] Bricelj A, Ng YLD, Ferber D, et al. Influence of Linker Attachment Points on the Stability and Neosubstrate Degradation of Cereblon Ligands. ACS Med Chem Lett. 2021;12(11):1733-1738. doi:10.1021/acsmedchemlett.1c00368.
[5] Atilaw Y, Poongavanam V, Svensson Nilsson C, et al. Solution Conformations Shed Light on PROTAC Cell Permeability. ACS Med Chem Lett. 2021;12(1):107-114. doi:10.1021/acsmedchemlett.0c00556.
[6] Zhang D, Ma B, Dragovich PS, et al. Tissue distribution and retention drives efficacy of rapidly clearing VHL-based PROTACs. Commun Med. 2024;4:87. doi:10.1038/s43856-024-00505-y.
扩展阅读:
1. PROTAC Linker连接臂设计全解析:三元复合物、细胞渗透、代谢稳定性与柔性刚性优化策略
2. 全球首个PROTAC药物vepdegestrant获批!ESR1突变ER+HER2-晚期乳腺癌新药与口服成药性深度解析
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论