

































2026年5月1日,美国食品药品监督管理局(FDA)批准了vepdegestrant(商品名VEPPANU)。其适应证为:经FDA授权检测确认存在ESR1突变、雌激素受体阳性(ER+)、人表皮生长因子受体2阴性(HER2−)的晚期或转移性乳腺癌成人患者,且疾病已在至少一线内分泌治疗后进展。它是FDA批准的首个PROTAC类异双功能蛋白降解剂。[1-2]
这一批准的意义需要准确界定:它不是人类第一次利用泛素—蛋白酶体系统治疗疾病,也不是第一种具有蛋白降解作用的药物;但它首次证明,经过理性设计的异双功能PROTAC可以完成从机制验证、口服暴露、临床获益到监管批准的完整转化链条。
不过,一款药物获批,并不意味着PROTAC的成药难题已经被普遍解决。相反,它更加清楚地说明:从“细胞中能够降解目标蛋白”到“患者能够长期、稳定、可控地用药”,中间还隔着溶解、吸收、分布、代谢、清除、组织暴露、三元复合物形成以及安全性等一整套相互牵制的问题。
传统小分子通常依靠占据靶蛋白的功能位点而发挥作用。PROTAC则由目标蛋白配体、E3泛素连接酶配体和连接臂(linker)组成,通过同时结合目标蛋白与E3连接酶,促进目标蛋白泛素化,随后由蛋白酶体降解。它属于“事件驱动”药理模式:药效取决于是否完成有效降解事件,而不只取决于对某个结合位点的持续占有。
但“事件驱动”不等于不需要充分暴露,也不等于每个PROTAC都能在体内高效循环使用。真实药效仍受细胞内游离浓度、三元复合物稳定性与协同性、泛素化效率、蛋白降解速度、目标蛋白再合成速度以及药物停留时间共同影响。
一、“五规则”并未失效,但不能被当成一票否决
Lipinski“五规则”原本是一组用于早期发现阶段的经验性风险提示:当化合物的氢键供体多于5个、氢键受体多于10个、分子量大于500 Da,或计算脂水分配系数cLogP大于5时,出现较差吸收或渗透的概率往往上升。[3] 需要注意,极性表面积和可旋转键数量并不属于Lipinski原始“五规则”,而是后来常与其配合使用的补充描述符。
完整PROTAC由两个配体和一段连接臂构成,分子量常处于700—1000 Da甚至更高,并可能同时具有较高极性、较多氢键位点和较大的构象自由度。因此,它们通常处于“超越五规则”(beyond Rule of Five,bRo5)的化学空间。
然而,“超出五规则”只意味着开发风险增加,并不等于必然不能口服。对这类分子,二维结构上的分子量、cLogP和拓扑极性表面积只能提供起点,不能替代对离子化状态、真实暴露极性、构象集合、外排转运、固态性质和体内清除的实验评价。
已有口服PROTAC研究表明,可口服性更像一个多参数窗口,而不是单一阈值。降低暴露的氢键供体、控制可旋转键数量、避免过度外排、减少非特异性结合并维持足够溶解度,往往比机械追求某一个“漂亮数字”更重要。[5-8]
二、口服暴露不是一个指标,而是一条连续链路
口服生物利用度可以粗略理解为三个环节的乘积:药物被肠道吸收的比例(Fa)、逃过肠壁代谢的比例(Fg)和逃过肝脏首过清除的比例(Fh)。任一环节接近零,最终进入体循环的母体药物都会非常有限。
因此,PROTAC口服成药性不应只被简化为“溶解度够不够”或“Caco-2渗透率高不高”。更完整的问题是:药物能否从制剂中释放并溶解,能否穿过肠上皮,是否受到P-gp等转运体外排,能否避开过快的肠壁和肝脏代谢,能否在血浆和组织中维持足够的非结合暴露,最后能否进入目标细胞并完成降解。
这条链路也解释了为什么体外降解活性很强的化合物,可能在动物体内几乎没有药效;也解释了为什么一个体外活性并非最强、但暴露和组织分布更均衡的化合物,反而更有机会成为临床候选物。
三、第一关不是“水里能溶”,而是“在真实胃肠环境中能形成可吸收剂量”
口服药必须先从固体制剂中释放,并在胃肠液中形成可供吸收的溶解态。PROTAC往往同时具有较大的疏水表面和多个极性基团,容易面临水相溶解度低、晶体堆积稳定以及溶出缓慢等问题。
这里需要纠正一个常见的过度简化:低溶解度并不只是“分子太疏水”的结果,也不能仅凭极性基团数量推断。晶型、盐型、无定形状态、晶格能、离子化、分子构象和介质组成都会改变最终溶解表现。开发中应区分热力学溶解度、动力学溶解度与制剂条件下的溶出行为。
普通缓冲液中的低溶解度,也不一定等同于胃肠道中的低溶解度。含胆盐、磷脂和脂肪酸成分的生物相关介质,可能通过胶束增溶提高某些高脂溶性分子的表观溶解度;但这种增溶是否真正转化为吸收,还取决于药物从胶束向肠上皮转移的能力。
食物可改变胆汁分泌、胃排空、胃肠pH、液体体积和脂质环境,因而可能显著改变暴露。VEPPANU的获批说明书要求随餐服用,这正是食物效应可以成为临床给药条件的一例,而不是对所有PROTAC都适用的通则。[2]
结构难以进一步压缩时,盐型筛选、共晶、无定形固体分散体、喷雾干燥、热熔挤出、自乳化体系或其他增溶技术可以扩大可吸收剂量。但制剂只能解决特定环节:若化合物同时具有极低溶解度、极低内在渗透性和强外排,仅靠制剂通常难以彻底补救。
四、第二关是渗透,但“低渗透”可能由多种机制叠加
PROTAC至少要跨越两类关键膜屏障:首先穿过肠上皮进入体循环,随后进入目标组织和目标细胞。较高分子量、较大的暴露极性、过多氢键供体以及过高柔性,都会增加被动跨膜的难度。
连接臂是调节渗透性的核心杠杆之一。长PEG链可能改善某些水相性质,却也可能增加极性表面积和构象自由度;短烷基链、环状单元、局部甲基化或更刚性的连接方式,有时能降低暴露极性和外排,但也可能损害溶解度或三元复合物几何构型。因此,连接臂优化不是简单的“越短越好”或“越刚越好”。[5,11]
PAMPA反映的是特定条件下的被动扩散,不能模拟细胞摄取、主动外排和细胞膜组成。Caco-2、MDCK或转染MDR1的细胞模型能够提供更多信息,但对PROTAC仍可能出现低回收率、强非特异性吸附和沉淀等问题。
含血清蛋白的接收液或低吸附耗材有时能改善质量平衡,却不能保证模型因此具有更好的体内预测性。2025年的一项系统研究显示,加入血清虽可降低非特异性结合、提高部分化合物回收率,但其Caco-2结果并未稳定预测口服吸收;因此必须同时报告质量平衡、表观渗透率、外排比和实验浓度下的溶解状态。[8]
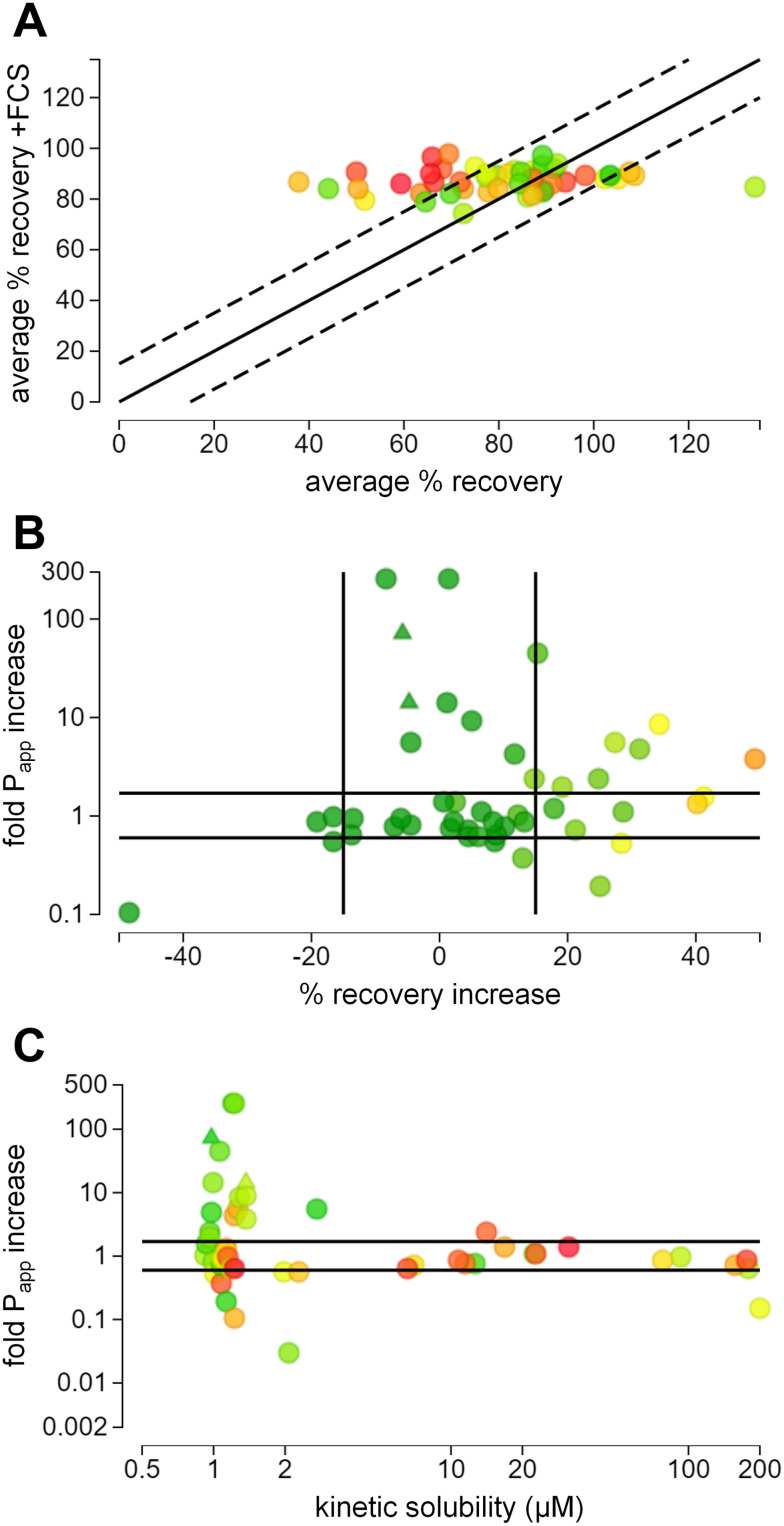
(A) 在Caco-2细胞培养中,添加和不添加10%FCS时的平均回收率。添加FCS后,回收率提高至80–100%。颜色编码表示clog D值:从≤1(绿色)经黄色到≥6(红色)。(B)Papp倍数变化与回收率变化的相关性。黑线代表这两个参数各自的95%置信区间。颜色编码表示标准条件下的平均回收率,颜色编码表示clog D值:从≤20%(红色)经黄色到≥80%(绿色)。(C)Papp倍数变化与动力学溶解度的关系。动力学溶解度>4μM 的 PROTAC© 的Papp值大多没有差异,而动力学溶解度<4μM 的 PROTAC© 的Papp值则存在显著差异。颜色编码表示堵塞程度 D:从≤1(绿色)经黄色到≥6(红色)。
对PROTAC而言,外排转运尤其值得前置评估。最新数据提示,在某些内部化合物集合中,外排比对口服吸收的区分能力可能强于部分“变色龙性”描述符。[9]
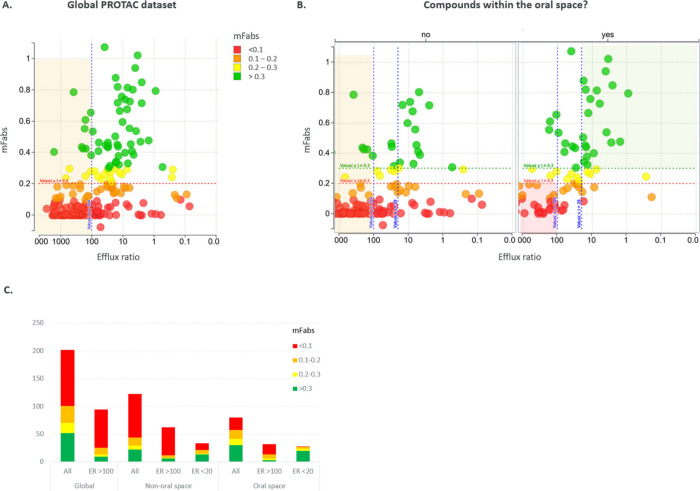
(A)外排比率与mFabs图,86%的ER > 100的化合物的mFabs < 20%;(B)在口腔空间内(右图,ChromlogD [3–7],ePSA [110–150]):当ER < 20时,化合物的mFabs > 0.3的概率较高;当ER > 100时,化合物的mFabs < 0.1的概率较高;(C)化合物根据其吸收分数在不同空间内的分布。
这并不否定构象的重要性,而是提醒研究者:只看被动渗透,容易漏掉真正限制体内吸收的转运因素。
五、“变色龙式”构象是重要解释框架,但不是万能通行证
PROTAC不是一张静止的二维结构图,而是会在不同环境中采样多种构象。在极性环境中,分子可能暴露更多极性基团以维持溶解;进入低极性环境时,部分分子可通过分子内氢键、芳香堆积和折叠构象遮蔽极性,从而降低瞬时暴露极性并改善被动跨膜。
这种环境依赖的性质变化常被称为“分子变色龙性”。它可能帮助解释为什么某些明显超出五规则的分子仍具有口服暴露,但并非所有PROTAC都具有足够的变色龙行为,也不是形成分子内氢键越多越好。过度折叠可能遮挡两个配体的结合构象,降低三元复合物形成效率。
设计上可以通过重新安排氢键供体与受体、改变连接位点、控制局部柔性、引入立体中心或局部甲基化来调节构象集合。验证则应结合溶液核磁共振、不同极性溶剂中的构象分析、分子动力学、色谱衍生的暴露极性指标以及细胞渗透实验,而不能只依赖单一计算构象。[6-7]
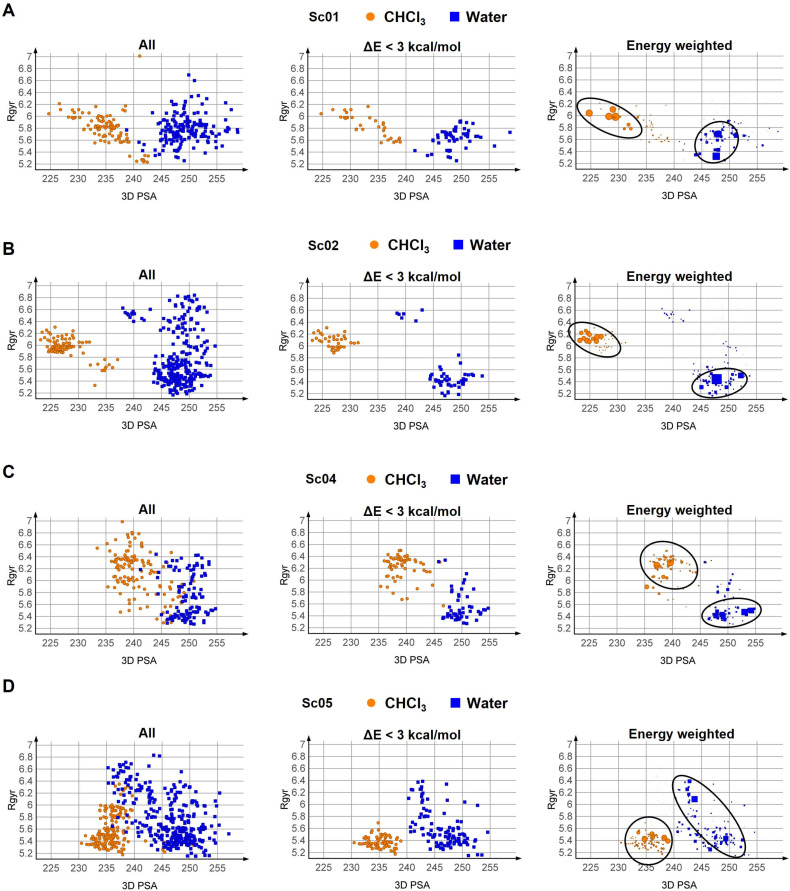
图示为采用不同方案获得的构象系综的 Rgyr 与 3D PSA 的关系图。三种构象采样方案与三种构象选择方案相结合。(A) Sc01,(B) Sc02,(C) Sc04,(D) Sc05。黑色圆圈标出了高概率构象组。Sc03 和 Sc06 未予展示,因为它们分别与 Sc02 和 Sc05 提供了相同的构象系综。
六、第三关是代谢与清除:完整PROTAC不是两个配体性质的简单相加
PROTAC进入体内后,可能在肠道、肝脏、血液或目标组织中被代谢。完整分子形成后,两个配体的空间暴露、电子环境和酶结合方式都会改变,因此不能用单独配体的稳定性简单推断完整PROTAC的稳定性。
常见代谢软点包括连接臂或配体上的氧化、N-去烷基化、O-去烷基化、脂肪链羟基化,以及酯、碳酸酯、部分氨基甲酸酯或其他易水解连接键的断裂。普通酰胺键通常相对稳定,不应笼统地与酯键并列为“常见快速水解位点”;但在特殊结构或特定酶环境下,酰胺水解仍可能发生。
较长、柔性的连接臂通常提供更多潜在代谢位点,但增加刚性或脂溶性也可能降低溶解度、增加组织滞留和非特异性结合。目标不是追求“完全不代谢”,而是把清除速度、组织暴露和安全边界调到适合给药方案的范围。
除CYP酶外,还应根据结构风险补充醛氧化酶、黄嘌呤氧化酶、肝胞质、肝S9组分、肝细胞以及血浆或全血稳定性研究。醛氧化酶和血液酶活性存在明显物种差异,动物中稳定并不保证在人源体系中同样稳定。
传统小分子IVIVE方法在PROTAC上也可能失真。高蛋白结合、高细胞孵育结合和低回收率会使内在清除率被低估或高估。已有研究表明,实测孵育体系非结合比例可能比通用预测公式更适合用于PROTAC清除率外推。[8]
七、E3配体、连接臂和目标配体必须作为一个整体优化
E3配体不仅决定招募哪一种E3连接酶,也会改变整个分子的分子量、极性、构象、代谢和组织分布。常见VHL配体往往比常见CRBN配体更大、更极性,但这只是常见化学型的总体趋势,不能据此得出“CRBN一定优于VHL”的结论。
E3选择至少要同时回答四个问题:目标细胞中是否有足够的E3复合物;目标蛋白与E3能否形成空间上有利的三元复合物;目标蛋白表面是否存在可被有效泛素化的赖氨酸与合适几何关系;所选E3配体是否带来额外的非目标蛋白降解、代谢或安全风险。
连接臂也不只是距离尺。其长度、刚性、出口向量和立体化学会影响三元复合物的协同性、停留时间和泛素化效率。目标配体亲和力较高并不自动意味着降解更强;一个亲和力中等但能形成高质量三元复合物的分子,可能具有更好的降解效能和选择性。[10]
因此,结构—活性关系应升级为结构—性质—降解关系:每次修改都要同步查看二元结合、三元复合物、DC50、Dmax、降解速率、蛋白恢复时间、细胞渗透、外排、溶解度和体内暴露,而不是只比较某一个终点。
八、蛋白结合与非特异性吸附:总浓度不等于可作用浓度
PROTAC常具有较高血浆蛋白结合。通常情况下,非结合药物是被动跨膜和即时靶点结合的直接驱动力,因此只比较血浆总浓度,可能高估真正可进入组织和细胞的药量。
但“只有游离药物有意义”也不应被绝对化。蛋白结合可能形成可逆储库,组织分配、细胞内滞留、结合动力学和目标蛋白周转都会影响药效持续时间。更合理的做法是同时分析总暴露、非结合暴露和靶组织暴露,并用药效数据检验哪一种指标最能解释蛋白降解。
非特异性吸附还会使样品黏附在塑料、玻璃、过滤膜和管路表面,造成表观低渗透、表观高代谢或虚假的低游离比例。实验中应采用低吸附材料、设置空白回收与质量平衡、检查沉淀,并避免把“检测不到”直接解释为“被代谢掉”。
九、剂量越高不一定降解越强,但Hook effect不能被机械套用
在适当浓度下,一个PROTAC分子可以同时结合目标蛋白和E3连接酶,形成有效三元复合物。浓度过高时,目标蛋白和E3可能分别被不同PROTAC分子占据,使有效三元复合物比例下降,这被称为Hook effect。
不过,Hook effect并非所有PROTAC、所有细胞和所有体内剂量都会明显出现。二元亲和力、三元复合物协同性、目标蛋白与E3浓度、细胞内游离药物浓度以及降解与再合成动力学都会改变其出现位置和幅度。高协同性体系可能把Hook effect推迟到更高浓度,甚至在实际给药范围内并不明显。[10]
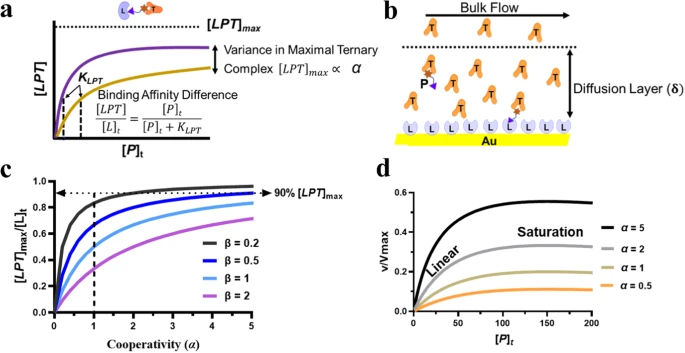
a:两种 PROTAC 结合亲和力和协同性差异的示意图。b:SPR 流动池示意图,靶蛋白 (T) 和 PROTAC (P) 位于连接酶功能化的芯片(L)上,扩散层厚度为δ。c:SPR 芯片上PROTAC 介导的三元复合物分数 ([LPT]max/[L]t) 与协同性 (α)的关系图,其中β值变化。虚线箭头指示90%的临界点。[LPT]max在SPR芯片上。d:表示在固定浓度下,通过改变α值,初始靶标泛素化速率与总PROTAC浓度[P]t的关系。[L]t,KLP和KTP。
因此,开发中不能只追求最高Cmax,也不能仅凭单点DC50判断剂量。更有价值的是建立血浆与靶组织暴露、非结合浓度、目标蛋白降解、蛋白恢复和功能药效之间的PK/PD关系。
事件驱动药理还可能出现“药物浓度已下降,但蛋白尚未恢复”的时间错位。给药频率应由蛋白再合成速度和药效持续时间共同决定,而不是机械地追随血浆半衰期。
十、制剂与前药可以扩展空间,但不能替代分子本身的可开发性
当主要瓶颈来自晶型或溶出时,制剂优化可能显著提高暴露;当主要问题是高暴露极性、强外排和低细胞渗透时,结构优化通常更关键;当母体药物快速清除时,则应优先处理代谢软点、转运和蛋白结合问题。
前药策略可以暂时遮蔽极性基团,改善吸收后再释放母体,但对PROTAC尤其复杂:新增基团会进一步增大分子,并引入前药稳定性、转化速率、组织选择性、中间体暴露和安全性等新的变量。现阶段,前药更适合作为明确机制假设下的专项方案,而不是口服失败后的通用补救。
同样,纳米递送、自乳化或复杂增溶体系也会增加制造、放大、稳定性和临床变异风险。最稳妥的策略通常是先把分子优化到具有基本可开发性,再让制剂解决剩余的固态和溶出问题。
十一、DMPK必须前置,并采用“可解释的失败”而不是“堆满数据”
PROTAC项目最危险的开发顺序,是先把细胞降解活性做到极致,再把少数化合物送去做药代。若一个系列从一开始就存在严重低溶解度、强外排、连接臂快速断裂或极高非特异性结合,继续围绕DC50做精细优化,往往只是在缺乏成药基础的骨架上反复修饰。
更合理的是建立分层DMPK级联:
- 早期发现阶段:快速测定离子化与脂溶性、动力学和生物相关介质溶解度、PAMPA或替代被动渗透指标、细胞渗透与外排比、血浆蛋白结合、孵育体系非结合比例、肝微粒体/肝细胞稳定性、血浆或全血稳定性以及样品回收率。其目的不是得到最完整的数据,而是尽快识别结构性失败模式。
- 先导优化阶段:围绕目标配体、连接位点、连接臂和E3配体建立结构—性质—降解关系。每轮修改同步比较三元复合物、细胞内降解、溶解度、渗透、外排、代谢软点和小动物静脉/口服PK;必要时鉴定断裂产物和主要代谢物,并确认其是否仍有药理活性或安全风险。
- 候选化合物阶段:确认口服生物利用度、剂量线性、食物影响、血浆与靶组织总/非结合暴露、目标蛋白降解起效与恢复、主要代谢物、药物相互作用风险、物种差异以及安全窗。只有把“给了多少药”与“组织中降解了多少蛋白、持续多久、产生什么功能效应”联系起来,才能判断临床剂量是否可行。
级联设计的核心不是测试项目越多越好,而是每个实验都能回答一个明确决策问题:失败来自溶解、被动渗透、外排、代谢、组织分布,还是三元复合物本身?只有失败机制可解释,结构优化才有方向。
除了内部实验数据,外部研发证据也可以帮助确定DMPK级联的优先顺序。例如,同靶点或同类降解机制的临床项目采用了何种剂型和给药方式,是否考察食物影响、剂量线性和药物相互作用,以及项目目前处于推进、调整还是终止状态,都可能为风险判断提供参考。研发团队可以利用摩熵医药数据库对相关药物、靶点、企业、临床试验、注册进展和专利信息进行关联检索,再将这些外部线索转化为需要优先验证的实验问题。
Vepdegestrant临床试验信息
数据库不能代替溶解度、渗透性、代谢稳定性和体内PK实验,但能够减少信息分散造成的判断盲区,让DMPK筛选更具有问题导向。
十二、从“能降解”走向“能成药”
PROTAC的价值在于,它把药物设计的目标从“持续抑制一个位点”扩展到“诱导一次可产生持续后果的蛋白降解事件”。这为部分缺乏经典活性口袋、仅需结合而不必直接抑制的靶点提供了新的干预机会。
但“可降解”不等于“可成药”,更不等于“可口服”。一个口服PROTAC必须同时在固态、胃肠环境、肠上皮、肝脏、血浆、目标组织和目标细胞中完成一系列彼此矛盾的任务:既要有足够溶解度,又要能够跨膜;既要足够稳定,又不能因过度脂溶而产生不可控蓄积;既要形成高质量三元复合物,又要避免非目标蛋白降解和过窄的安全窗。
Lipinski“五规则”并没有在PROTAC时代失效。它仍是一块有用的风险路标,只是不能再被当成终点。真正决定成功的,是能否把二维结构描述符、动态构象、体外ADME、体内PK、组织分布和降解PK/PD整合为同一条证据链。
因此,最有希望走向患者的PROTAC,未必是体外DC50最低的那个,而更可能是那个在降解效能、暴露、选择性、可制造性和安全性之间取得可重复平衡的分子。
参考资料:
1. U.S. Food and Drug Administration. FDA approves vepdegestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer. 2026-05-01.
2. VEPPANU (vepdegestrant) tablets, U.S. Prescribing Information. Initial U.S. Approval: 2026.
3. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3-26. doi:10.1016/S0169-409X(96)00423-1.
4. Hamilton EP, et al. Vepdegestrant, a PROTAC Estrogen Receptor Degrader, in Advanced Breast Cancer. N Engl J Med. 2025. doi:10.1056/NEJMoa2505725.
5. Klein VG, et al. Understanding and Improving the Membrane Permeability of VH032-Based PROTACs. ACS Med Chem Lett. 2020;11:1732-1738. doi:10.1021/acsmedchemlett.0c00265.
6. Structural and Physicochemical Features of Oral PROTACs. J Med Chem. 2024. doi:10.1021/acs.jmedchem.4c01017.
7. Conformational Sampling Deciphers the Chameleonic Properties of a VHL-Based Degrader. Pharmaceutics. 2023;15:272. PMID:36678900.
8. Maurer CK, et al. In vitro and in vivo ADME of heterobifunctional degraders: a tailored approach to optimize DMPK properties of PROTACs. RSC Med Chem. 2025;16:1746-1757. doi:10.1039/D4MD00854E.
9. Evaluation of Oral PROTAC Guidelines: Efflux Ratio Outweighs Chameleonicity Descriptors. ACS Med Chem Lett. 2026. doi:10.1021/acsmedchemlett.6c00043.
10. Affinity and cooperativity modulate ternary complex formation to drive targeted protein degradation. Nat Commun. 2023. doi:10.1038/s41467-023-39904-5.
11. Zhang J, et al. Structural Feature Analyzation Strategies toward Discovery of Orally Bioavailable PROTACs of Bruton's Tyrosine Kinase for the Treatment of Lymphoma. J Med Chem. 2022;65:9096-9125. doi:10.1021/acs.jmedchem.2c00324.
12. Evaluation of Cereblon-Directing Warheads for the Development of Orally Bioavailable PROTACs. J Med Chem. 2025. doi:10.1021/acs.jmedchem.4c02709.
扩展阅读:
1. 药物代谢全解析:CYP450酶作用机制+AI-PBPK模型,2026精准用药指南更新
2. 2026年全球双特异性抗体核心管线深度解析:依沃西单抗OS碾压PD-1,双抗ADC强势崛起
3. BE豁免实战指南:基于BCS分类与溶出曲线f2相似因子,NMPA/FDA一致性评价降本增效策略
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论