


































1.4.1 本周全球TOP10创新药研发进展

(1)康哲药业芦可替尼乳膏申报新适应症,治疗特应性皮炎
2月24日,康哲药业宣布 磷酸芦可替尼乳膏 的新适应症上市申请获得国家药监局受理,用于其他外用药控制不佳或不建议使用时,非免疫功能受损的2岁及以上儿童和成人轻中度特应性皮炎的局部短期和非持续性慢性治疗。此外,该申请因“符合儿童生理特征的儿童用药品新品种、剂型和规格”,已被纳入优先审评品种名单。
中国特应性皮炎III期研究显示,治疗8周后,磷酸芦可替尼乳膏 组达到研究者整体评估(IGA)评分为0或1分且较基线改善≥2分的受试者比例显著高于安慰剂组(63.0% vs 9.2%,P<0.001)。此外,治疗8周后,磷酸芦可替尼乳膏 组达到湿疹面积及严重程度指数评分较基线至少改善75%(EASI 75)的受试者比例亦显著优于安慰剂(78.0% vs 15.4%,P<0.001)。在该研究中,磷酸芦可替尼乳膏 组患者在治疗期出现的不良事件(TEAE)的严重程度大多数为轻度或中度,未发生导致研究药物用药终止的TEAE,整体安全耐受性良好。
(2)康弘药业抗失眠症1类新药获批临床
2月25日,中国国家药监局药品审评中心(CDE)官网公示显示,康弘药业申报的1类新药 KHN707片 获批临床,拟开发治疗失眠症。
根据康弘药业公告介绍,这是该公司自主研发的选择性食欲素2受体(OX2R)拮抗剂,属于化药1类创新药,剂型为片剂。前期已完成的研究结果显示 KHN707片 安全性较好,且在失眠模型中具有良好的治疗作用,预期临床应用前景较好。
公开资料显示,食欲素(orexin)是一种神经递质,通常被认为是睡眠-觉醒周期的主要调节剂。OX2R拮抗剂通过阻止食欲素与受体结合,从而抑制食欲素系统驱动的觉醒信号,代表了一种通过靶向调节觉醒系统来治疗失眠的新策略。
(3)GSK口服靶向新药利奈昔巴特片在中国申报上市
2月25日,中国国家药监局药品审评中心(CDE)官网公示显示,葛兰素史克(GSK)申报的1类新药 利奈昔巴特片 上市申请已获得受理。这是是GSK在研的回肠胆汁酸转运蛋白(IBAT)抑制剂linerixibat。根据CDE官网,该产品此前已经被纳入优先审评,适用于治疗原发性胆汁性胆管炎(PBC)成人患者的胆汁淤积性瘙痒。
PBC作为一种胆汁淤积性肝病,会导致肝脏胆汁流动受阻。由此引发的循环胆汁酸过量被认为是胆汁淤积性瘙痒症(一种无法通过抓挠缓解的内源性瘙痒)的致病因素。Linerixibat 是一种IBAT口服抑制剂,具有治疗与PBC相关的胆汁淤积性瘙痒的潜力。通过抑制胆汁酸重吸收,linerixibat 可减少循环中多种瘙痒介质。美国FDA和欧洲药品管理局(EMA)已授予 linerixibat 用于治疗与PBC相关的胆汁淤积性瘙痒的孤儿药资格。该产品的新药申请(NDA)已经于2025年6月获FDA及EMA受理,适应症为治疗原发性胆汁性胆管炎(PBC)成人患者的胆汁淤积性瘙痒。
(4)康方生物抗IL-4Rα单抗1类新药申报上市
2月25日,中国国家药监局药品审评中心(CDE)官网公示显示,康方生物申报的1类新药 曼多奇单抗注射液 上市申请获得受理。公开资料显示,曼多奇单抗(研发代号:AK120)是康方生物自主研发的新型人源化抗IL-4Rα(白介素-4受体α)单克隆抗体。
2025年8月,康方生物宣布 曼多奇单抗 在治疗中重度特应性皮炎(AD)的关键注册性3期临床研究中取得阳性结果,研究的主要终点、关键次要终点及其他预设的多项次要终点全部成功,具有统计学显著性和临床意义的改善。曼多奇单抗 显著改善了患者的皮损状态的同时,在早期瘙痒改善中疗效优异。根据康方生物此前新闻稿介绍,该公司正有序推进该适应症的上市申请(NDA)进程。
(5)勃林格殷格翰的宗艾替尼获得FDA特批,针对初治非小细胞肺癌
2月26日,FDA通过特批通道批准 宗艾替尼(zongertinib)用于治疗存在HER2(ERBB2)酪氨酸激酶结构域激活突变的不可切除或转移性非鳞状非小细胞肺癌(NSCLC)成人患者。宗艾替尼 由勃林格殷格翰开发,是全球首个且目前唯一获批的口服HER2酪氨酸激酶抑制剂(TKI),可选择性抑制HER2(ERBB2),同时避免对野生型EGFR的抑制,从而最大程度减少相关毒性。
2025年8月,宗艾替尼 相继获得FDA和国家药监局(NMPA)批准上市,用于治疗存在HER2(ERBB2)酪氨酸激酶结构域激活突变且既往接受过系统性治疗的不可切除或转移性非鳞状NSCLC成人患者。此次批准后,宗艾替尼 的适用人群进一步扩大至未接受过系统性治疗的人群。
(6)GSK全球首创乙肝新药在日本申报上市
2月26日,葛兰素史克(GSK)宣布慢性乙肝新药 Bepirovirsen 在日本递交上市申请。GSK认为,Bepirovirsen 有望成为首款实现慢性乙肝功能性治愈的药物。
Bepirovirsen 是GSK从Ionis引进的一种反义寡核苷酸(ASO)疗法,旨在通过抑制乙型肝炎病毒DNA的复制,进而抑制血液中乙型肝炎表面抗原(HBsAg)水平,并刺激免疫系统产生持久应答。该药物是慢性乙肝领域首款完成III期研究的小核酸药物。
此次上市申请是基于两项III期研究(B-Well 1和B-Well 2)的积极结果。这两项研究评估了 Bepirovirsen 对比安慰剂在接受过核苷类似物治疗且基线HBsAg不超过3000IU/ml的慢性乙肝患者中实现功能性治愈的有效性、安全性、药代动力学特征及持久性。结果显示,两项研究均达到了主要终点,即 Bepirovirsen 组患者实现功能性治愈的比例显著高于安慰剂组。此外,研究也达到了所有终点,并且 Bepirovirsen 在基础HBsAg不超过1000IU/ml的患者中的效果更为显著。
(7)赛诺菲宣布度普利尤单抗在中国获批两项新适应症
2月27日,赛诺菲宣布 度普利尤单抗注射液(商品名:达必妥)获国家药品监督管理局(NMPA)正式批准两项新适应症:用于治疗大疱性类天疱疮(bullous pemphigoid, BP)成人患者,以及用于6岁及以上儿童哮喘患者的维持治疗。此次双适应症获批标志着达必妥成为中国首个且目前唯一获批用于治疗大疱性类天疱疮的靶向生物制剂,以及中国首个且目前唯一覆盖6岁及以上儿童哮喘患者的抗IL-4Ra单抗,为这两类患者带来突破性的创新治疗选择。
度普利尤单抗 是一种全人源单克隆抗体,可抑制白细胞介素-4(IL-4)和白细胞介素-13(1L-13)通路的信号转导,而不是免疫抑制剂。在 度普利尤单抗 开发计划的多项III期临床试验中,该药表现出显著的临床获益,可抑制2型炎症,并证实IL-4和IL-13是2型炎症的关键核心驱动因素,在多种相关的常见并发疾病中发挥重要作用。
(8)亚虹医药光动力治疗产品上市申请获欧洲药品管理局受理
2月27日,亚虹医药宣布其APL-1702(CEVIRA)拟用于治疗宫颈高级别鳞状上皮内病变(HSIL)患者的上市许可申请已获得欧洲药品管理局(EMA)受理。
APL-1702 是一款光动力治疗产品,由 盐酸氨酮戊酸己酯软膏 和一次性使用宫颈光动力治疗灯组成,通过创新的局部给药与阴道内置冷光源设计,实现了治疗模式的突破。
(1)门诊“即治即走”便捷式治疗:其药械一体化设计,由妇科医生门诊操作,无需麻醉,将单次诊疗时间缩短至10分钟以内;放置后可立即恢复正常工作和生活,患者无需在医院等待,治疗完成后患者可自行取出装置。该“门诊短时放置+居家完成治疗”的模式,将极大提升医疗效率和治疗可及性。
(2)冷光源技术提升治疗舒适度:APL-1702采用的创新冷光源设计,可将治疗区域组织温度控制在42℃以内。临床数据显示,97%患者报告宫颈治疗区域无痛感,且无宫颈结构损伤记录,避免传统光动力治疗可能导致的组织热损伤,减少局部刺激,改善治疗舒适度。
(9)正大天晴JAK/ROCK抑制剂罗伐昔替尼片获批上市
2月28日,NMPA官网显示,正大天晴的1类创新药 罗伐昔替尼片(商品名:安煦)获批上市,适用于中危-2或高危的原发性骨髓纤维化(PMF)、真性红细胞增多症后骨髓纤维化(PPV-MF)或原发性血小板增多症后骨髓纤维化(PET-MF)成人患者的一线治疗,治疗疾病相关脾肿大或疾病相关症状。
罗伐昔替尼(TQ05105)是正大天晴自主研发的一款first-in-class、全新的口服小分子JAK/ROCK抑制剂。体外试验结果显示,该药能够有效抑制JAK家族激酶活性及ROCK激酶活性,能显著抑制细胞中STAT3和STAT5的磷酸化水平,从而抑制JAK/STAT信号通路传导作用,进而发挥抗肿瘤活性。
(10)济民可信KRAS G12C抑制剂获批上市
2月28日,国家药监局(NMPA)官网显示,济民可信子公司浙江杭煜制药的1类创新药KRAS G12C抑制剂 硫酸索西美雷塞片(商品名:济乐美)获批上市,适用于至少接受过一种系统性治疗的KRAS G12C突变型的晚期非小细胞肺癌(NSCLC)成人患者。
硫酸索西美雷塞片 此前已获国家药品监督管理局突破性治疗药物认定,其关键注册临床数据表现突出,在晚期NSCLC患者后线治疗中展现出优异的疗效与安全性:客观缓解率(ORR)高达55.2%,12个月总生存率(OS)达到60.4%,相较于同靶点已上市疗法,济乐美 不仅在生存获益上具备明显竞争优势,更采用“每日一次、一次两片”的便捷给药方案,耐受性优异,有效解决了患者长期用药的依从性痛点,展现了该产品在细分领域极具竞争力的差异化优势。
1.4.2 本周全球TOP10积极/失败临床结果


(1)百利天恒EGFR×HER3双抗ADC三阴性乳腺癌III期成功
2月23日,百利天恒宣布其自主研发的全球首创(First-in-class)、新概念(New concept)且唯一进入III期临床阶段的EGFR×HER3双抗ADC(iza-bren),在一项针对局部晚期或转移性三阴性乳腺癌(TNBC)的III期临床试验(研究编号:BL-B01D1-307)中,其预设的期中分析成功达到了PFS(无进展生存期)与OS(总生存期)双主要研究终点。
该III期临床研究独立数据监查委员会(iDMC)建议:“基于现有的分析结果,与监管沟通提前申报,同时继续对受试者进行随访”。顶线(topline)数据显示,iza-bren显著延长了PFS和OS,达到PFS和OS的双主要终点。这是 iza-bren 作为全球首创双抗ADC药物,在乳腺癌领域取得的首个III期研究阳性结果;同时,它也是 iza-bren 第3项达到主要研究终点的III期临床研究。适应症为既往经紫杉烷类治疗失败的不可手术切除的局部晚期或转移性三阴性乳腺癌。
(2)诺和诺德CagriSema头对头替尔泊肽减重III期研究未达主要终点
2月23日,诺和诺德宣布 CagriSema 对比 替尔泊肽 的减重III期REDEFINE 4研究未达到主要终点。CagriSema 为诺和诺德开发的一款复方药物,包含长效胰淀素(amylin)类似物Cagrilintide与GLP-1受体激动剂 司美格鲁肽。
REDEFINE 4研究是一项为期84周的开放标签临床试验(n=809),评估了 CagriSema(2.4mg+2.4mg)对比 替尔泊肽(15mg)治疗至少伴有一种肥胖相关合并症的肥胖患者的有效性和安全性。患者基线平均体重为114.2kg。若按治疗效果(即所有患者坚持治疗)评估,治疗84周后,CagriSema 组和 替尔泊肽 组患者的体重分别减轻了23.0%和25.5%。若按治疗方案(即无论患者是否坚持治疗)评估,治疗84周后,CagriSema 组和 替尔泊肽 组患者的体重分别减轻了20.2%和23.6%。总而言之,该研究未达到非劣效性主要终点。
(3)QurAlis公司公布其在研药物QRL-201 1/2期临床积极数据
2月23日,QurAlis公司宣布,其在研药物 QRL-201 在1/2期概念验证临床试验ANQUR中取得积极的中期数据。QRL-201 是一种潜在“first-in-class”的强效ASO疗法,旨在恢复肌萎缩侧索硬化(ALS)患者体内因TDP-43功能异常而下调的关键蛋白STATHMIN-2(STMN2)的表达,从而改变疾病进程。
ANQUR研究是一项双盲、安慰剂对照研究,共纳入69名患者。数据显示,QRL-201 在脊髓和运动皮层广泛分布,成功实现靶点结合;与自然病程对照组相比,患者STMN2蛋白水平显著升高(超过预设治疗阈值),并有效纠正了STMN2的异常剪接。同时,低剂量组患者的磷酸化神经丝重链(pNfH)的降低具有统计学显著性及临床意义。在散发性ALS患者中,QRL-201 整体呈现延缓ALSFRS-R评分下降的趋势,尤其在关乎患者生活自理能力的粗大运动子评分上表现突出。
(4)联邦制药与诺和诺德公布多肽疗法2期试验积极结果
2月24日,联邦制药国际控股有限公司(联邦制药)与诺和诺德联合发布了多肽疗法 UBT251 的中国2期临床研究的主要结果。UBT251 由联邦制药的全资附属公司联邦生物与诺和诺德共同开发,是一种长效合成肽类GLP-1(胰高血糖素样肽-1)、GIP(葡萄糖依赖性促胰岛素多肽)和GCG(胰高血糖素)三靶点受体激动剂。
本次公布结果的临床研究由联邦生物开展,旨在研究中国超重或肥胖患者每周一次注射2 mg、4 mg和6 mg剂量的UBT251与安慰剂对照的安全性和有效性。患者基线平均体重为92.2 kg,治疗24周后,UBT251 治疗组的平均体重降幅最高达19.7%(-17.5 kg),而安慰剂组仅为2.0%(-1.6 kg)。此外,与安慰剂组相比,UBT251 所有剂量组在关键次要终点方面均显示出统计学显著改善,包括腰围、血糖、血压和血脂。本次研究中,UBT251 显示出良好的安全性和耐受性。最常见的不良事件为胃肠道反应,绝大多数为轻度至中度,且随时间推移而减轻,这与基于肠促胰素的疗法一致。基于该研究结果,联邦制药计划启动一项针对中国超重或肥胖患者的3期临床研究。
(5)Monte Rosa Therapeutics公布抗癌分子胶降解剂1/2期积极临床结果
2月25日,Monte Rosa Therapeutics公布了一项进行中的1/2期临床研究的最新临床数据。该研究评估其在研分子胶降解剂 MRT-2359 联合 恩扎卢胺(enzalutamide),在既往接受多线治疗的转移性去势抵抗性前列腺癌(mCRPC)患者中的疗效。分析显示,携带雄激素受体(AR)突变患者的靶病灶均观察到缩小,疾病控制率(DCR)达100%。公司计划于2026年第三季度启动一项新的验证性2期研究,针对携带AR突变的患者开展评估。
MRT-2359 是一款高效、高选择性、口服生物利用度良好的在研GSPT1靶向分子胶降解剂。包括前列腺癌在内的MYC驱动型肿瘤依赖增强的致癌蛋白翻译以支持快速生长。MRT-2359 通过选择性降解翻译终止因子GSPT1以破坏蛋白翻译过程,从而产生治疗效果。MRT-2359 治疗可降低多种与前列腺癌相关的致癌蛋白水平,包括AR、MYC及细胞周期蛋白D1-E2F,并在多种mCRPC临床前模型中显示出显著的抗肿瘤活性。
(6)礼来RET激酶抑制剂3期研究取得积极结果,针对肺癌
2月26日,礼来公司宣布,LIBRETTO-432 3期临床研究取得阳性顶线结果。该研究评估了 塞普替尼 作为辅助治疗相较于安慰剂的有效性和安全性。研究达到了其主要终点,证实在研究者评估的早期(II-IIIA期)转染重排(RET)融合阳性非小细胞肺癌患者中,塞普替尼 带来了高度统计学显著且具有临床意义的无事件生存期改善。
塞普替尼(原代号LOXO-292)是一种高选择性、强效的RET激酶抑制剂,并具有中枢神经系统(CNS)活性。该产品可能同时作用于肿瘤细胞和正常细胞,从而导致不良反应。RET驱动基因改变通常与其他致癌驱动基因改变互斥。在中国,塞普替尼由礼来研发,信达生物负责在中国大陆商业化。
(7)礼来口服GLP-1小分子药物orforglipron临床3期结果登《柳叶刀》
2月26日,礼来(Eli Lilly and Company)公布了ACHIEVE-3研究的详细结果。这是一项为期52周的头对头3期临床试验,旨在比较每日一次口服GLP-1受体激动剂小分子 orforglipron 与口服司美格鲁肽(semaglutide)在二甲双胍治疗控制不佳的2型糖尿病成人患者中的安全性与疗效。该研究共纳入1698名受试者,设置四个治疗组,分别为12 mg与36 mg orforglipron,以及7 mg与14 mg口服司美格鲁肽。相关研究成果已发表于《柳叶刀》期刊。
研究结果显示,在主要终点及所有关键次要终点上,orforglipron 整体表现优于活性对照组,在降低糖化血红蛋白(A1C)及减重方面均实现更显著改善。在主要终点方面,高剂量 orforglipron 组的A1C降低幅度达到2.2个百分点,显著优于高剂量活性对照药物组的1.4个百分点。在关键次要终点中,接受高剂量 orforglipron 治疗的患者平均减重19.7磅(9.2%),相比高剂量活性对照药物组的11.0磅(5.3%),相对减重幅度提高73.6%。此外,orforglipron 还在多项心血管风险指标方面较基线实现具有临床意义的改善,包括非高密度脂蛋白胆固醇(non-HDL-C)、高密度脂蛋白胆固醇(HDL-C)、极低密度脂蛋白胆固醇(VLDL-C)、总胆固醇、收缩压及甘油三酯水平。
(8)映恩生物公布B7H3靶向ADC新药1/2期新临床数据
2月27日,映恩生物宣布在2026年美国临床肿瘤学会泌尿生殖系统肿瘤研讨会(ASCO GU)上以海报形式公布了与BioNTech合作开发的靶向B7H3的抗体偶联药物(ADC)DB-1311/BNT324 在转移性去势抵抗性前列腺癌(mCRPC)患者中的最新临床研究数据。本次报告的数据来自一项全球多中心1/2期临床研究。
截至2025年12月29日,共纳入146例既往接受过多线治疗的mCRPC患者,中位年龄为70岁,中位既往治疗线数为4线。其中,52例患者(超总人群1/3)既往接受过³⁷⁷Lu-PSMA-617放射配体治疗(Lu-177),该亚组人群中位既往治疗线数为5线,87%既往接受过紫杉烷类治疗,40%既往同时接受过多西他赛和卡巴他赛。疗效方面,DB-1311 在经多线治疗的mCRPC患者中展现出令人鼓舞的抗肿瘤活性。129例疗效可评估的患者中,中位影像学无进展生存期(rPFS)为11.3个月,中位总生存期(OS)为22.5个月。亚组分析显示,84例既往未接受过Lu 177的患者中位rPFS为13.6个月;45例既往接受过Lu 177的患者疗效与整体人群相当,中位rPFS同样为11.3个月,中位OS尚未达到。安全性方面,DB-1311/BNT324 延续既往的优异表现,未发生新的安全性信号。
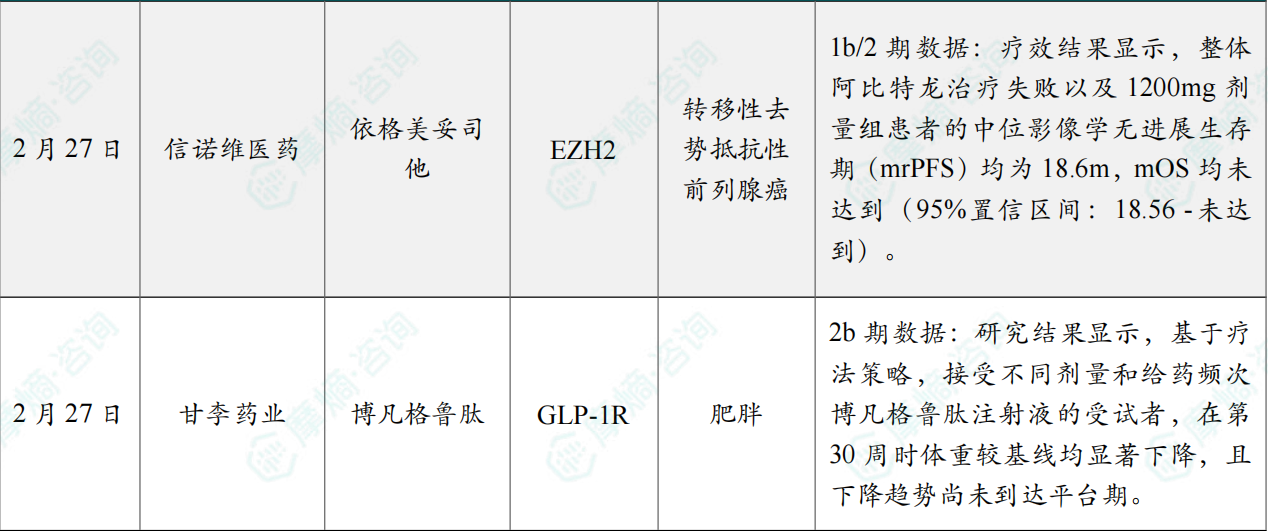
(9)信诺维公布EZH2抑制剂依格美妥司他治疗前列腺癌1b/2期临床最新数据
2月27日,信诺维医药宣布其自主研发的在研1类新药EZH2抑制剂——依格美妥司他(XNW5004)的最新临床数据在2026年美国临床肿瘤学会泌尿生殖系统肿瘤研讨会(ASCO GU)上公布。研究表明,依格美妥司他 联合 恩扎卢胺 在既往接受过阿比特龙治疗的转移性去势抵抗性前列腺癌(mCRPC)治疗中展现出良好的疗效及安全性。
在本次公布的 依格美妥司他 联合 恩扎卢胺 治疗mCRPC的Ib/II期临床研究中,截至2026年1月28日,共入组87例既往接受过新型内分泌治疗的mCRPC患者,整体安全性和耐受性良好,无DLT事件发生,最终选择Igermetostat 1200mg, BID作为临床II期推荐剂量(RP2D)。研究纳入56例既往接受过阿比特龙治疗的患者进行疗效分析,中位随访时间为17.5个月。疗效结果显示,整体阿比特龙治疗失败以及1200mg剂量组患者的中位影像学无进展生存期(mrPFS)均为18.6m,mOS均未达到(95%置信区间:18.56 -未达到)。在1200mg剂量下,12个月rPFS率为71.8%。在16例基线存在可测量病灶的患者中,最佳总体缓解率为25.0%,其中包含4例部分缓解。35例前列腺特异性抗原(PSA)可评估患者中,11例(31.4%)达到PSA较基线下降≥50%。
(10)甘李药业博凡格鲁肽II期临床研究成果在Nature出版社顶刊发表
2月27日,甘李药业自主研发的1类新药胰高血糖素样肽-1受体激动剂(GLP-1RA)双周制剂 博凡格鲁肽(GZR18)注射液,在中国肥胖/超重患者中完成的IIb期临床研究结果正式发表于国际权威期刊《Signal Transduction and Targeted Therapy》。
该研究是一项随机、双盲、安慰剂对照的IIb期临床研究,在中国20个研究中心开展。研究结果显示,基于疗法策略,接受不同剂量和给药频次 博凡格鲁肽注射液 的受试者,在第30周时体重较基线均显著下降,且下降趋势尚未到达平台期。
主要结果如下:在 博凡格鲁肽注射液 Q2W各剂量组中,受试者体重较基线变化百分比呈剂量依赖性下降:12 mg、18 mg、24 mg和48 mg组分别下降9.75%、12.55%、13.66%和16.09%;24 mg QW组下降16.69%,而安慰剂组下降1.15%。48 mg Q2W与24 mg QW组间的减重效果无显著差异(单侧检验P>0.025),表明每两周给药一次在降低注射频率、提升治疗便捷性的同时,仍可保持与每周给药相当的疗效。博凡格鲁肽 治疗各剂量组减重达标率均显著高于安慰剂组,其中48 mg Q2W组体重减轻幅度≥5%、10%、15%和20%的受试者比例分别为96.8%、82.7%、61.5%和39.2%。
同期事件:
1. 2026年第9周02.23-03.01国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2026年第9周02.23-03.01国内仿制药/生物类似物申报/审批数据分析
3. 2026年第9周02.23-03.01国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2026.02.23-2026.03.01) },如需查看或下载完整版报告,可点击!
扩展阅读:
1. 2026年第7-8周02.09-02.22全球创新药研发概览
2. 2026年第6周02.02-02.08全球创新药研发概览
3. 2026年第5周01.26-02.01全球创新药研发概览
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论