

































1.4.1 本周全球TOP10创新药研发进展
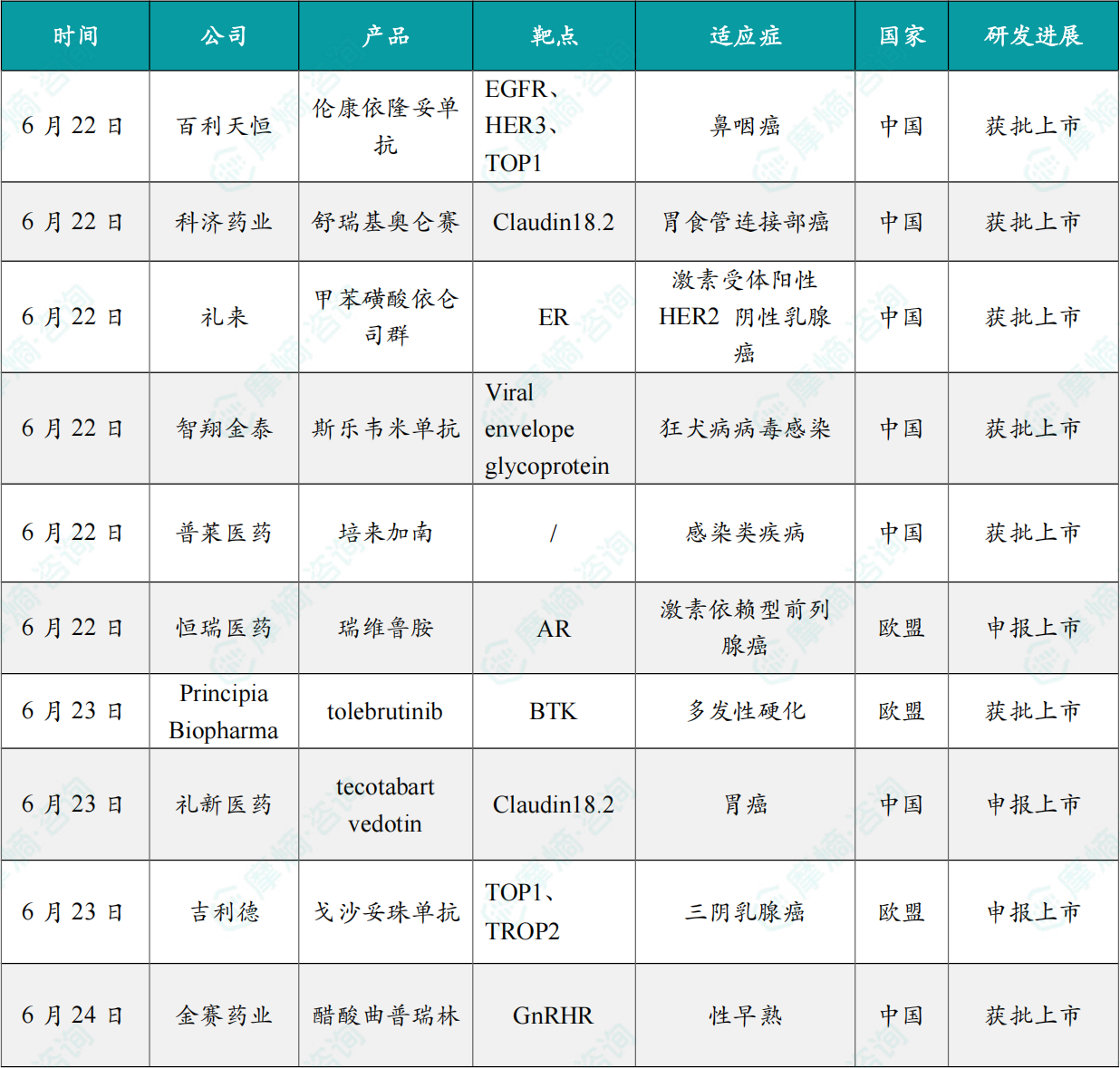
(1)百利天恒双抗ADC在华获批上市
6月22日,国家药监局(NMPA)官网显示,注射用BL-B01D1(伦康依隆妥单抗)获批上市,用于既往经PD-1/PD-L1单抗治疗且经至少两线化疗(至少一线含铂)治疗失败的复发性或转移性鼻咽癌患者。
2025年7月,伦康依隆妥单抗治疗鼻咽癌的III期BL-B01D1-303研究在期中分析达到主要终点,成为全球首个完成III期研究的双抗ADC。结果显示,伦康依隆妥单抗组和对照组患者的ORR分别为54.6%和27.0%。
伦康依隆妥单抗是百利天恒自主研发的全球首创(First-in-class)、新概念(New concept)的EGFR/HER3双抗ADC,2023年12月,BMS与百利天恒达成协议,以84亿美元的总交易额获得该药物在中国和美国以外地区的独家开发和商业化权益以及在美国的合作开发和商业化权益。
(2)科济药业CLDN18.2 CAR-T疗法在华获批上市
6月22日,国家药监局(NMPA)网站显示,科济药业的CLDN18.2 CAR-T疗法舒瑞基奥仑赛获批上市,用于治疗既往接受过至少二线治疗失败的CLDN18.2阳性晚期胃或食管胃结合部腺癌。
I/II期CT041-ST-01研究(n=156)结果显示,舒瑞基奥仑赛组患者的中位无进展生存期(mPFS)较医生选择治疗方案组(包括紫杉醇、多西他赛、伊立替康、阿帕替尼或纳武利尤单抗)显著延长(3.25 vs 1.77个月,HR=0.37,P<0.0001)。
(3)礼来口服SERD药物在华获批上市
6月22日,国家药监局(NMPA)官网显示,礼来的口服雌激素受体拮抗剂 依仑司群(imlunestrant)获批上市,用于治疗雌激素受体阳性(ER+)、人表皮生长因子受体2阴性(HER2-)、携带ESR1突变的晚期或转移性乳腺癌成人患者,这些患者的疾病在至少接受过一线内分泌疗法后出现进展。
在III期EMBER-3研究中,与内分泌疗法相比,imlunestrant将疾病进展或死亡风险降低了38%。在携带ESR1突变的乳腺癌患者中,与氟维司群或依西美坦组相比,imlunestrant组患者的无进展生存期(PFS)显著改善(5.5个月 vs 3.8个月,HR=0.62,p=0.0008)。
(4)智翔金泰狂犬病病毒双特异抗体在华获批上市
6月22日,国家药品监督管理局(NMPA)官网显示,智翔金泰的 斯乐韦米单抗注射液(GR1801)获批上市,用于成人疑似狂犬病病毒暴露后的被动免疫。斯乐韦米单抗是全球首个用于狂犬病被动免疫的双特异性抗体。
斯乐韦米单抗是一款由智翔金泰自主研发的重组全人源抗狂犬病病毒(Rabies Virus,RABV)双特异性抗体,注册分类为治疗用生物制品1类,作用靶点为RABV的包膜糖蛋白(Glycoprotein,G蛋白)。斯乐韦米单抗分子设计满足WHO关于抗狂犬病病毒抗体开发的建议——采用针对不同抗原位点的多株单抗组合成“鸡尾酒式”组合制剂,以保证对不同病毒株或病毒的不同基因型的有效性。
(5)普莱医药多肽类抗菌药在华获批上市
6月22日,NMPA 官网显示,普莱医药申报的 1 类新药「培来加南」在国内获批上市。这是一款多肽类抗菌药物,本次获批适应症为:治疗由金黄色葡萄球菌、表皮葡萄球菌、铜绿假单胞菌、溶血葡萄球菌、鲍曼不动杆菌等导致的浅表性继发性创面感染,包括烧烫伤创面感染、物理性损伤创面感染等。
培来加南喷雾剂(PL-5)是一款 First-in-class 的加南类抗感染药物,由普莱医药自主研发,拥有全球知识产权。该药抗菌谱广,对多种耐药菌包括超级细菌 MRSA(耐甲氧西林金黄色葡萄球菌)及含 NDM-1 基因多重耐药鲍曼不动杆菌等均具有非常强的杀菌优势,曾获国家「十二五」及「十三五」「重大新药创制」科技重大专项支持。
(6)恒瑞首个创新药在欧盟申报上市
6月22日,恒瑞医药宣布瑞维鲁胺片的上市许可申请在近日获得欧洲药品管理局(EMA)受理,用于联合雄激素剥夺治疗(ADT),治疗高瘤负荷的转移性激素敏感性前列腺癌(mHSPC)成年男性患者。这是恒瑞医药首个自主研发的创新药在欧盟申报上市。
瑞维鲁胺片是第二代雄激素受体(AR)抑制剂,相较于第一代AR抑制剂,具有更强的AR 抑制作用,且无激动作用。此次申报上市得到了III期SHR-3680-III-HSPC(CHART)研究的支持。研究结果显示,瑞维鲁胺联合ADT一线治疗高瘤负荷mHSPC具有显著的生存获益和可耐受的安全性。截至2022年2月28日,相较于对照组,瑞维鲁胺组患者发生影像学进展或死亡的风险降低54%(中位rPFS:NR vs 23.5个月,HR=0.46)。瑞维鲁胺组患者的OS显著延长,死亡风险降低了42%(中位OS:NR vs NR,HR=0.58,p=0.0001)。
(7)赛诺菲第二款自免BTK抑制剂在欧盟获批上市
6月23日,赛诺菲宣布 Tolebrutinib(商品名:Cenrifki)获得欧洲药品管理局(EMA)批准上市,用于治疗无复发情况的继发进展型多发性硬化症(SPMS)。该药物是首款可用于治疗非复发性SPMS的药物,也是赛诺菲旗下第二款获批上市的自免BTK抑制剂。
赛诺菲的第一款自免BTK抑制剂 Rilzabrutinib(商品名:Wayrilz)已于2025年在美国和欧洲获批上市。此次批准得到了III期HERCULES研究的支持。该研究纳入了1131例非复发性SPMS患者。结果显示,中位随访133周时,Tolebrutinib(60mg,每日1次)组患者确认出现持续至少6个月的残疾进展的比例显著低于安慰剂组(22.6% vs 30.7%,HR=0.69,P=0.003)。
(8)中国生物制药CLDN18.2 ADC申报上市
6月23日,CDE网站显示,中国生物制药收购所得的CLDN18.2 ADC 维特柯妥拜单抗(Tecotabart vedotin,LM-302)申报上市,用于至少接受过两种系统治疗的CLDN18.2阳性、局部晚期或转移性胃或胃食管交界处腺癌患者。
维特柯妥拜单抗是礼新医药基于其独家多次跨膜蛋白抗体发现平台开发的CLDN18.2特异性抗体、可裂解连接子以及毒素载荷甲基澳瑞他汀E(MMAE)组成的一款CLDN18.2 ADC。2025年7月,中国生物制药以9.51亿美元的价格全资收购礼新医药,获得了该药物的所有权益。I/II期LM302-01-102研究数据显示,CLDN18.2阳性胃癌患者接受维特柯妥拜单抗(1.8mg/kg,每2周1次)治疗后,客观缓解率(ORR)为32.7%,中位无进展生存期(PFS)为4.9个月,总生存期(OS)为10.9个月。
(9)吉利德ADC在欧盟获批
6月23日,吉利德科学公司(Gilead Sciences)宣布,欧盟委员会(EC)已批准 Trodelvy(sacituzumab govitecan)作为单药疗法,用于治疗既往未接受过转移性疾病系统治疗、且不适合接受PD-1或PD-L1抑制剂治疗的不可切除或转移性三阴性乳腺癌(TNBC)成人患者。
根据新闻稿,Trodelvy 是首款获EC批准用于一线转移性TNBC的抗体偶联药物(ADC),也是欧洲20年来针对特定一线转移性TNBC患者获批的首个新治疗选择。
此次批准主要基于3期ASCENT-03研究数据。该研究显示,与标准化疗相比,Trodelvy 作为一线治疗在无进展生存期(PFS)方面达到高度统计学显著且具有临床意义的改善;在不适合接受PD-1/PD-L1抑制剂治疗的患者中,Trodelvy 将疾病进展或死亡风险降低38%。
(10)金赛药业注射用醋酸曲普瑞林获批上市
6月24日,金赛药业宣布其研发的2.2类新药 注射用醋酸曲普瑞林(II)于近日获得NMPA批准上市,用于体重20kg及以上儿童中枢性性早熟的治疗。促性腺激素释放激素激动剂是国内外指南和共识推荐的中枢性性早熟治疗药物。在此基础上,金赛药业研发的 注射用醋酸曲普瑞林(II)是一款1个月缓释制剂,属于化学药品2.2类。
本次获批是基于一项在儿童中枢性性早熟中开展的多中心、开放、单臂研究。注射用醋酸曲普瑞林(II)在该研究中表现出了良好的有效性和安全性。新闻稿指出,该产品是国内首个完成中枢性性早熟前瞻性注册研究的1个月促性腺激素释放激素激动剂(GnRHa)缓释剂型,为性早熟患儿提供了新的治疗选择。
1.4.2 本周全球TOP8积极/失败临床结果
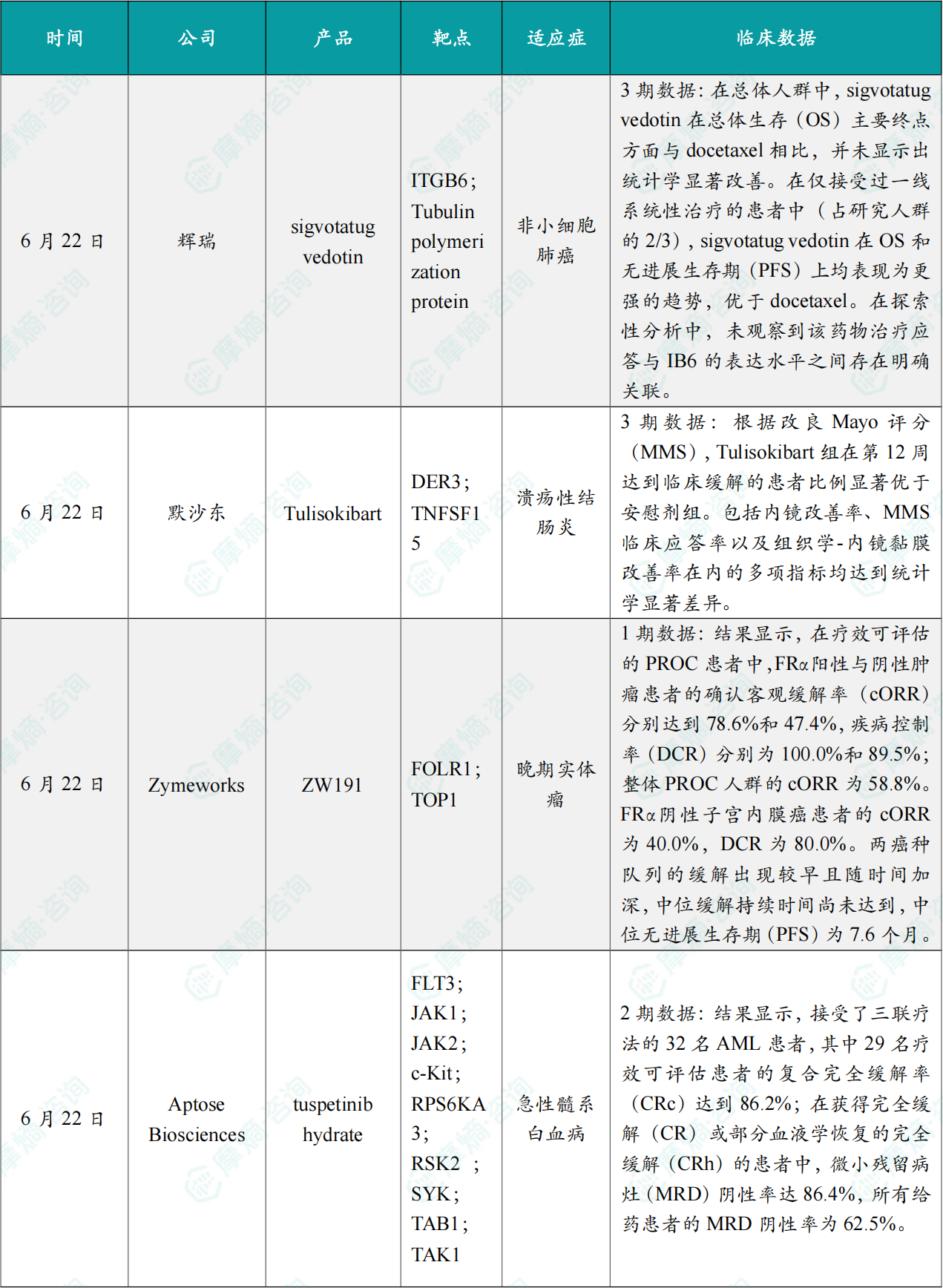
(1)辉瑞ADC肺癌3期临床失败
6月22日,辉瑞公布靶向整合素β6(IB6)的抗体偶联药物(ADC)sigvotatug vedotin 关键3期SigVie-002研究顶线数据,这款药物是辉瑞2023年斥资430亿美元收购Seagen获得的核心管线资产,用于经治晚期非鳞非小细胞肺癌(NSCLC)的后线治疗,但试验未达成预设主要终点总生存期(OS)显著获益目标,消息公布后辉瑞盘后股价下跌超1%。
整体人群数据分析显示,相较于多西他赛,sigvotatug vedotin未能带来具有统计学意义的总生存改善,未达到研究核心终点;药物整体安全性可控,不良反应特征与既往早期研究数据保持一致。不过分层分析释放出积极信号:占全部受试者三分之二、仅接受过一线系统治疗的二线患者亚组中,sigvotatug vedotin在总生存期、无进展生存期(PFS)两项关键疗效指标上均展现出明显获益趋势。
(2)默沙东溃疡性结肠炎临床III期成功
6月22日,默沙东宣布,其在研新药 Tulisokibart(MK-7240)在针对中度至重度活动性溃疡性结肠炎(UC)的Ⅲ期ATLAS-UC诱导研究中,成功达到了主要终点和关键次要终点。在第12周时,该药物成功让患者在MMS标准下实现了临床缓解,并且没有发现任何安全隐患。这一成就让 Tulisokibart 成为全球首个在Ⅲ期临床试验中证明能在12周内实现临床缓解的抗TL1A单克隆抗体。默沙东计划在即将召开的科学大会上公布详细数据,并与监管机构分享这些振奋人心的结果。
(3)Zymeworks公布ZW191的1期临床试验的新数据
6月22日,Zymeworks公司公布了其ADC疗法 ZW191 的1期临床试验新数据。ZW191 靶向叶酸受体α(FRα),该靶点在约75%的高级别浆液性卵巢癌和约70%的肺腺癌中表达。ZW191 具有独特的设计,可高效进入FRα阳性细胞,并释放Zymeworks开发的具旁观者效应的载荷——拓扑异构酶I抑制剂 ZD06519。
此次公布的结果显示,ZW191 在铂类耐药卵巢癌及子宫内膜癌患者中展现出显著的抗肿瘤活性。截至2026年3月9日的数据,在疗效可评估的PROC患者中,FRα阳性与阴性肿瘤患者的确认客观缓解率(cORR)分别达到78.6%和47.4%,疾病控制率(DCR)分别为100.0%和89.5%;整体PROC人群的cORR为58.8%。FRα阴性子宫内膜癌患者的cORR为40.0%,DCR为80.0%。两癌种队列的缓解出现较早且随时间加深,中位缓解持续时间尚未达到,中位无进展生存期(PFS)为7.6个月。此外,该药物具备良好的耐受性特征和较宽的治疗窗口,支持继续开展临床开发。
(4)Aptose Biosciences公布1/2期TUSCANY试验的联合治疗试验数据
6月22日,Aptose Biosciences公司公布了其针对新确诊的急性髓系白血病患者开展的1/2期TUSCANY试验的新数据。这些患者接受了不同剂量的 tuspetinib 联用标准剂量的 venetoclax 和 azacitidine。该三联疗法正在被开发作为一种安全且不依赖于特定突变的一线治疗方案,用于治疗那些无法接受诱导化疗的新确诊AML患者,这些患者具有多样的突变。
结果显示,32名AML患者接受了TUS+VEN+AZA三联疗法,其中29名疗效可评估患者的复合完全缓解率(CRc)达到86.2%;在获得完全缓解(CR)或部分血液学恢复的完全缓解(CRh)的患者中,微小残留病灶(MRD)阴性率达86.4%,所有给药患者的MRD阴性率为62.5%。具有不同遗传特征(包括TP53突变、复杂核型和未突变FLT3)的AML患者,在安全实现治疗缓解的同时,也达到了MRD阴性。安全性方面,tuspetinib 与标准剂量的VEN/AZA联用耐受性良好,未发生与治疗相关的死亡、QTc间期延长、CPK升高或分化综合征,且其药代动力学特性未受联合用药、抗真菌药或食物的显著影响。
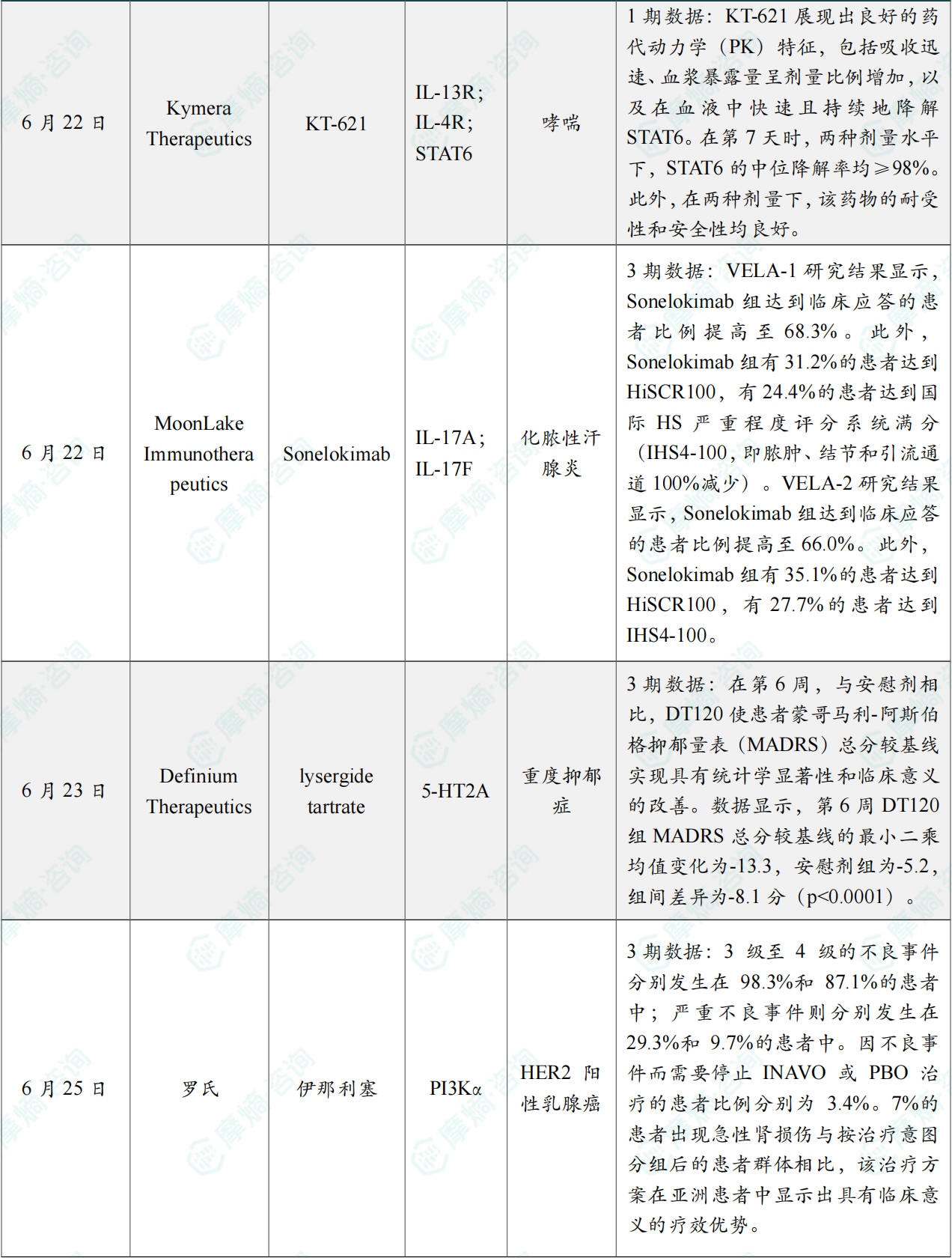
(5)Kymera Therapeutics公司公布KT-621的1期临床试验数据结果积极
6月22日,Kymera Therapeutics公司公布了其在研口服、潜在“first-in-class”的STAT6降解剂 KT-621 在健康日本成年人中进行的1期临床研究结果。该公司此前已报告KT-621在非日本健康志愿者及中重度特应性皮炎患者中的1期临床数据。在这些研究中,KT-621 使受试者血液和皮肤中的STAT6快速、深度且持续地降解,并显著降低疾病相关2型炎症生物标志物,特应性皮炎以及共病哮喘和过敏性鼻炎的临床终点和患者报告结局显著改善,安全性良好。
此次公布的结果显示,KT-621 展现出良好的药代动力学(PK)特征,包括吸收迅速、血浆暴露量呈剂量比例增加,以及在血液中快速且持续地降解STAT6。在第7天时,两种剂量水平下,STAT6的中位降解率均≥98%。此外,在两种剂量下,该药物的耐受性和安全性均良好。总体而言,这些在日本健康成人中获得的结果与在非日本成人及特应性皮炎患者中观察到的结果相当,进一步支持了 KT-621 作为慢性2型炎症性疾病新型口服疗法的潜力。
(6)默克公布Sonelokimab治疗化脓性汗腺炎的两项III期试验数据
6月22日,Moonlake公布IL-17A/F纳米双抗 Sonelokimab 治疗化脓性汗腺炎三期临床VELA 52周数据。
数据表明,治疗52周,67.2%的患者达到HiSCR75,33.1%的患者达到HiSCR100(VELA-1:68.3% HiSCR75, 31.2% HiSCR100; VELA-2: 66.0% HiSCR75, 35.1% HiSCR100)。治疗52周,26.0%的患者达到IHS4-100(反映炎症完全缓解),VELA-1为24.4%,VELA-2为27.7%。去年9月披露的中期数据,Sonelokimab 治疗16周的HiSCR分别为为17.3%、10.3%,HiSCR50分别为21.3%、15.7%。此次更新的长期数据表明,疗效持续深化。
(7)Definium Therapeutics宣布其抑郁症创新疗法3期结果积极
6月23日,Definium Therapeutics今日宣布,其在研药物 DT120(lysergide)口腔崩解片(ODT)在名为Emerge的3期临床研究中取得积极顶线结果。该研究是一项随机、双盲、安慰剂对照试验,旨在评估单剂量100 µg DT120 ODT用于治疗成人抑郁症(MDD)的疗效。
研究达到主要终点:在第6周,与安慰剂相比,DT120 使患者蒙哥马利-阿斯伯格抑郁量表(MADRS)总分较基线实现具有统计学显著性和临床意义的改善。数据显示,第6周 DT120 组MADRS总分较基线的最小二乘均值变化为-13.3,安慰剂组为-5.2,组间差异为-8.1分(p<0.0001)。除达到主要终点外,DT120 的疗效还表现出快速且持久的特点。第1周时,该药相较安慰剂的MADRS总分最小二乘均值降幅为14.2分(p<0.0001);至第12周时,相较安慰剂的降幅仍为7.3分(p<0.0001)。安全性方面,DT120 ODT总体耐受性良好,治疗期间出现的不良事件中,99%为轻度至中度、短暂性事件,且主要发生在给药当天。研究未发现新的安全性信号,包括未观察到自杀意念或行为增加;两组停药率均较低且相当。DT120 是一种麦角碱衍生物,属于经典血清素能致幻剂,可作为5-羟色胺2A(5-HT2A)受体部分激动剂发挥作用。
(8)罗氏公布了其INAVO120的3期临床试验亚洲患者亚组分析的结果
6月25日,罗氏公司公布了其针对接受 伊那利塞(INAVO)/安慰剂(PBO)联合 哌柏西利(PALBO)、氟维司群(FULV)治疗的、携带PIK3CA突变、激素受体阳性、人表皮生长因子受体2阴性(HER2-)内分泌耐药晚期乳腺癌(aBC)亚洲患者(pts)的亚组分析。
结果显示,与按治疗意图分组后的患者群体相比,INAVO + PALBO + FULV 方案在亚洲患者中显示出具有临床意义的疗效优势。该方案没有带来新的安全问题,且与按治疗意图分组后的患者群体相比,INAVO 方案的停药率和剂量调整率也更低。这些数据进一步证明了 INAVO120 方案在 PIK3CA 突变、激素受体阳性、HER2 阴性、对内分泌治疗有抵抗性的晚期乳腺癌患者中的疗效优势。
同期事件:
1. 2026年第26周06.22-06.28国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2026年第26周06.22-06.28国内仿制药/生物类似物申报/审批数据分析
3. 2026年第26周06.22-06.28国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2026.06.22-2026.06.28) },如需查看或下载完整版报告,可点击!
查数据,找摩熵!想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论