


































1.口服给药与BCS分类系统
小分子新药还是多以口服给药为主,而口服给药中又以固体制剂居多。2014年,美国食品药品监督管理局批准的新药数量为41个(CDER,2014年报告),不同给药途径的比较显示,19个(46%)新药批准以胶囊剂或片剂的形式口服。口服给药自早期以来一直是最重要的给药途径。近年来,由于通过注射给药途径的兴起,这种明显的主导地位发生了变化。然而,口服制剂很可能在未来继续发挥关键作用,因为它们易于管理、稳定性好、生产工艺成熟、商品成本低。
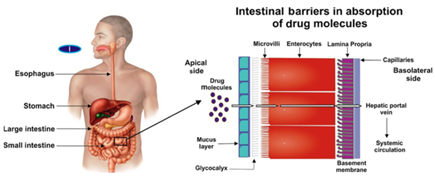
图1 药物吸收过程中存在的各种胃肠道屏障的示意图。(来源于参考文献1)
但是口服给药途径胃肠道,能否完成胃肠道的征途达到血液循环,进而到达作用靶器官靶组织靶细胞将是药物发挥疗效的重要前提。图1左图为药物吸收过程中存在的各种胃肠道屏障的示意图。图1右图代表了固制剂通过的各种器官。大多数药物是通过小肠吸收的。存在几种肠道屏障,药物分子通过这些屏障到达系统循环;各种屏障包括粘液层、糖被(glycocalyx)、微绒毛、肠细胞、固有层和细胞毛细血管。
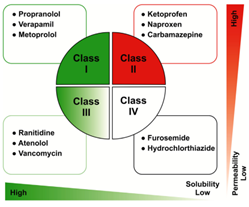
图2 生物药剂学分类系统(BCS)示例。(来源于参考文献1)
为了更好的理解药物的吸收,提出了基于药物溶解性和渗透性的生物药剂学分类系统(BCS分类系统)。药物的生物药剂学分类系统是根据药物的溶解性和渗透性进行分类的。如果药物符合某些标准,就可以获得生物等效性豁免。根据该分类系统,药物分为四类,即I类、II类、III类和IV类。I类为高溶解性和高渗透性,II类为低溶解性和高渗透性,III类为高溶解性和低渗透性,IV类为低溶解度和低渗透性。药物溶解度低是生物药剂学分类系统的II类和IV类药物的共同特性。药物渗透性低是生物药剂学分类系统的Ⅲ类和IV类药物的共同特性。
2.口服吸收障碍与增溶技术
由于药物发现和开发过程中溶解度低的化合物越来越多,用于提高口服药物吸收和生物利用度的使能技术的数量也在增加,各种使能技术如图3所示,通过对这些障碍的认识,有可能使用使能技术提高溶解度和溶出率的主要手段合理化—即通过降低药物晶格能、增加可用药物表面积或增加溶剂化能量。例如,脂质、表面活性剂和助溶剂会增加GI流体的疏水性微相(如囊泡和胶束)的体积和特性,从而增加难溶性药物的溶解度,进而提高化合物的体内暴露量。但是对于如何提高药物的因生物屏障带来的暴露问题的制剂技术少之又少。
药物口服给药吸收的障碍总结如下:
a.生物利用度的物理化学障碍
•高晶格能,通常随着化合物熔化温度(Tm)的升高而增加;
•水溶剂化的低能量,通常随着化合物的LogP值增加而降低(即亲油性)——通常称为“油脂球”化合物;
•两者的结合,其中高晶体能量对溶解度的影响因低溶剂化能量(通常称为“砖尘”化合物)而加剧。
b.生物利用度的生物障碍
•吸收的药物回流到肠腔(通常是P-gp或BCRP转运蛋白介导),
•肠道中的系统前药物代谢(主要通过细胞色素P450酶),以及
•广泛的肝脏首过药物代谢
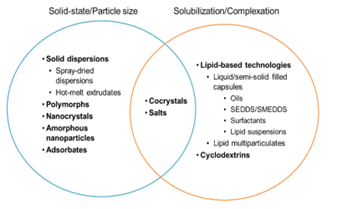
图 3 简图说明了各种使能技术提高药物溶解度/溶出率的主要机制,从而可能以更低的成本实现更快、更成功的开发,从而改善口服吸收。(来源于参考文献2)
3.基于P-gp外排的抑制提高药物体内暴露
前面我们提高基于BCS分类系统,BCSⅢ、Ⅳ类化合物口服面临的体内暴露问题,其实就渗透性问题。提高化合物的体内渗透性最有效的办法一是化合物修饰改变化合物的结构特征(相当于全新NCE),二是在药物体内递送的过程中使用渗透促进剂。使用渗透促进剂相较于提高化合物因溶解度问题带来的困扰更加难以对付。因为渗透性提高技术的效果不好(因胃肠道液体的稀释作用而发挥的作用常常不佳;可能造成组织损伤和不良反应)且变异性大。目前,FDA尚未批准任何辅料作为吸收促进剂。今天,我们简单介绍一个基于P-gp外排造成化合物体内渗透性不佳,借助TPGS促进体内暴露的案例。
P-糖蛋白(P-gp)是一种膜结合转运蛋白,介导多种结构无关的药物和其他外源性物质的主动转运。它沿肠道全长表达,也在肝脏(小管膜)、肾、血脑屏障、和胎盘。肠P-gp位于上皮细胞的顶膜上。利用ATP水解产生的能量,P-gp驱动各种底物逆浓度梯度外排,从而降低其细胞内浓度,在药物的情况下降低其口服生物利用度(图4)。这种复合糖蛋白(分子量为170kDa;图4)是MDR1基因的产物,并且在没有修饰的情况下外排药物,从而赋予多药耐药性。
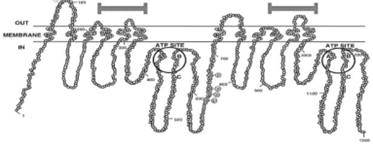
图4 P-gp结构示意图。(来源于参考文献3)
除了抗癌和抗HIV药物外,用于其他疾病适应症的NCEs也是P-gp底物,人们对抑制P-gp从而提高这些分子的生物利用度非常感兴趣。讨论和比较有效抑制P-gp的制剂策略。这些策略是独立的和组合的:(a)联合给药另一种P-gp底物/特异性抑制剂,和(b)在制剂中掺入非特异性脂质和/或聚合物。第一种方法虽然在抑制P-gp方面非常有效,但在制剂中使用了第二种活性化合物,因此对这种组合产品施加了监管限制和漫长的开发时间。辅料抑制剂似乎具有最小的非特异性药理学活性,因此可以避免特定活性化合物抑制剂的潜在副作用。
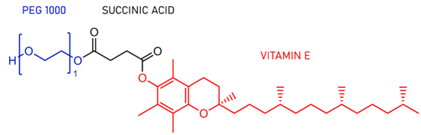
图5TPGS结构示意图。(来源于参考文献4)
D-ɑ-生育酚聚乙二醇琥珀酸酯(维生素ETPGS或TPGS)已被FDA批准为安全佐剂,并广泛用于药物输送系统。TPGS为淡黄色的蜡状固体近乎无味,能溶于水,也能溶于乙醇等大多数极性有机溶剂。TPGS熔点约39℃且有较高的降解温度,在200℃下稳定。室温时临界胶束浓度的质量分数约为0.02%,亲水亲油平衡值(HLB)约为13~17。由于TPGS的两亲性质及良好的水溶性,对亲脂性物质而言是很好的非离子性表面活性剂。当其与难溶性药物形成胶束或乳剂时,将显著增加药物在胃肠中的吸收,从而提高生物利用度。有报道,TPGS对P-gp有抑制作用,转运受P-gp阻滞的药物(如阿霉素、紫杉醇、长春碱、秋水仙碱等)与TPGS合用后,都明显增加了在胃肠道内的吸收,提高了生物利用度。TPGS的生物和理化特性为其在药物递送中的应用提供了多种优势,如高生物相容性,增强药物溶解性,改善药物渗透性和选择性抗肿瘤活性。值得注意的是,TPGS可以抑制ATP依赖性P-gp的活性,并作为克服肿瘤中多重耐药性(MDR)的有效赋形剂。
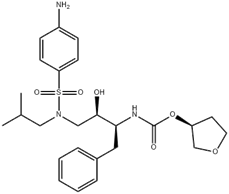
图6.安普那韦(MW=505.63)结构式。(来源于参考文献3)
安普那韦对酶的抑制活性Ki=0.6nmol·L−1(HIV1)和19nmol·L−1(HIV2),抑制HIV感染细胞的活性IC50=40nmol·L−1。水溶性S=190μg·mL−1。由于有良好的药效学和药动学性质,GSK公司将其进入开发阶段,命名为安普那韦(amprenavir),经临床研究于1999年FDA批准上市。CMC为0.2mg/mL的TPGS也显示出改善安普那韦的溶解度和肠道通透性,据报道,TPGS的作用是:(a)通过胶束增溶提高溶解度(S),从而提高安普韦的溶出,(b)增强安普那韦穿过肠壁的渗透性(Peff),可能是通过药物外排抑制。在一项特定的研究中,TPGS增强了药物的总吸收通量(J=Peff*S)(图6),通过增加其溶解度和肠道通透性。尽管TPGS主要通过抑制P-gp而被广泛用作难溶性药物的药物溶剂和口服生物利用度增强剂,在某些情况下,TPGS对P-gp抑制和药物吸收的影响很小或没有影响。有关报道,也表明体外作用与体内作用的差异,体外用量和体内用量的差异,包括对于不同的化合物分子,似乎也需要进行筛选和试验。
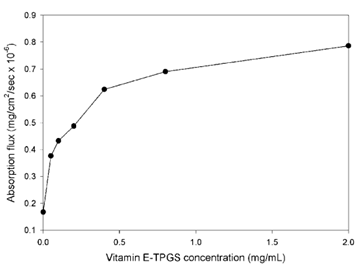
图7 TPGS存在下的安普那韦吸收。吸收通量水平高于0.5mg/mL的TPGS,表明单体TPGS抑制P-gp。(来源于参考文献3)
医药行业仍旧具有未曾攀爬的高山,而到达高山之巅,注定充满着荆棘。面对可开发性较差的药物分子,如何更好的递送到病灶,达到药到病除的目的,可谓任重道远。我相信随着竞争压力的增加促使研发投入的增加,科技科学的进步,路永远在脚下,光明的未来将是不久的明天!
推荐阅读:科研新突破!DNA纳米实现精准血栓治疗,药物递送技术新型解码
参考文献:
1.Physicochemical, Pharmaceutical, and Biological Considerations in GIT Absorption of Drugs
2. Technology Selection for Bioavailability Enhancement
3. Lipid Formulation Strategies for Enhancing Intestinal Transport and Absorption of P-Glycoprotein (P-gp) Substrate Drugs: In vitro/In vivo Case Studies
4. Recent Advances in the Application of Vitamin E TPGS for Drug Delivery
5. The use of TPGS in drug delivery systems to overcome biological barriers
6. 聚乙二醇1000维生素 E 琥珀酸酯应用进展
7.基于结构设计的蛋白酶抑制剂沙奎那韦、安普那韦和地瑞那韦
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!





















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论